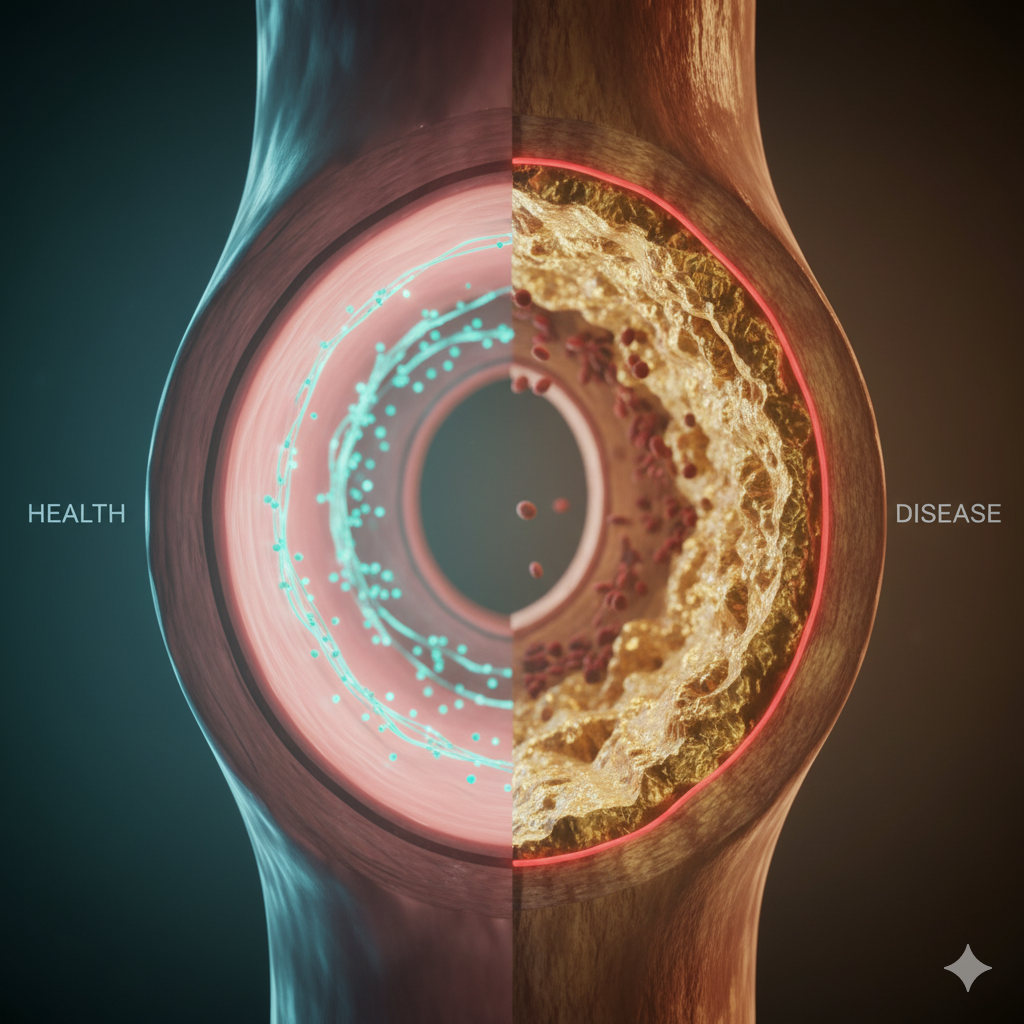
بیماری عروق کرونر (CAD)
نقشه یادگیری این مقاله
۱. پادکست: برای درک کلی، ابتدا به پادکست گوش دهید.
۲. ویدیو: ویدیو آموزشی را برای یادگیری عمیق مشاهده کنید.
۳. مطالعه متن: در نهایت، متن مقاله را به عنوان منبع جامع مرور کنید.
مشاهده ویدیو در آپارات
بیماری عروق کرونر (CAD): ریشهیابی و کالبدشکافی انسداد شریانهای قلب
تحلیل جامع پاتوفیزیولوژی، تشخیص و درمان
۱. مقدمه و تعریف جامع: فراتر از یک انسداد مکانیکی
بیماری عروق کرونر (Coronary Artery Disease – CAD) که در متون پزشکی نوین اغلب با عنوان کلیتر “بیماری قلبی ایسکمیک” شناخته میشود، یک پدیده پاتوفیزیولوژیک چندوجهی است که نمیتوان آن را صرفاً به یک انسداد مکانیکی ساده در لولههای خونرسان قلب تقلیل داد. این بیماری، که همچنان علت اصلی مرگومیر در جوامع صنعتی و در حال توسعه است، نتیجه نهایی یک فرآیند التهابی مزمن، پیشرونده و سیستمیک به نام آترواسکلروز (Atherosclerosis) است. در حالی که تصور عمومی بر این است که CAD به معنای رسوب چربی در سطح داخلی رگهاست، نگاه دقیق علمی نشان میدهد که این بیماری یک نبرد بیولوژیک پیچیده در لایههای عمیق دیواره شریان است که دههها قبل از بروز اولین علامت بالینی آغاز میشود.
آترواسکلروز فرآیندی است که در آن سد دفاعی عروق شکسته شده، لیپوپروتئینها به دام میافتند و سیستم ایمنی بدن علیه بافت خودی وارد عمل میشود. این بیماری با باریک شدن یا انسداد لومن (مجرا) شریانهای کرونر مشخص میشود، اما پاتولوژی آن شامل تغییرات ساختاری عمیق در لایههای اینتیما (Intima) و مدیا (Media) دیواره رگ است. یکی از مفاهیم کلیدی که درک آن برای فهم ماهیت موذیانه این بیماری ضروری است، «پدیده گلاگوف» (Glagov Phenomenon) است. بر اساس این پدیده، شریانهای کرونر در مراحل اولیه تشکیل پلاک، دچار «بازسازی مثبت» (Positive Remodeling) میشوند؛ به این معنا که دیواره خارجی رگ منبسط میشود تا لومن رگ باز بماند و جریان خون حفظ شود. این مکانیسم جبرانی باعث میشود که پلاک آترواسکلروتیک بتواند حجم عظیمی از دیواره رگ (تا ۴۰ درصد سطح مقطع) را اشغال کند بدون اینکه در آنژیوگرافی استاندارد تنگی قابل توجهی دیده شود یا بیمار دچار علائم شود. بنابراین، CAD تنها بیماری تنگی رگ نیست، بلکه بیماری دیواره رگ است که ممکن است سالها به صورت خاموش و بدون علامت پیشرفت کند تا زمانی که ظرفیت جبرانی رگ به پایان برسد یا پلاک به صورت ناگهانی پاره شود.
۲. مکانیسم مولکولی و سلولی آترواسکلروز: کالبدشکافی فرآیند
درک عمیق CAD نیازمند بررسی دقیق مراحل تشکیل پلاک آترواسکلروتیک است. تحقیقات گسترده نشان میدهند که این فرآیند یک رسوبگذاری غیرفعال چربی نیست، بلکه یک پاسخ التهابی فعال به آسیب است که میتوان آن را در چندین مرحله متوالی و درهمتنیده تشریح کرد.
۲.۱. اختلال عملکرد اندوتلیال (Endothelial Dysfunction): آغاز ماجرا و نقش تنش برشی
سلامت سیستم قلبی-عروقی به یکپارچگی و عملکرد صحیح لایه تکسلولی پوشاننده داخل عروق، یعنی «اندوتلیوم»، وابسته است. اندوتلیوم سالم یک سطح غیرترومبوژنیک (ضد لخته) است که با ترشح مداوم نیتریک اکساید (NO) و پروستاسیکلین، تونوس عروقی را تنظیم کرده و از چسبیدن گلبولهای سفید و پلاکتها جلوگیری میکند. شروع آترواسکلروز با «اختلال عملکرد اندوتلیال» کلید میخورد، جایی که تعادل بین عوامل گشادکننده و تنگکننده عروق به هم میخورد و نفوذپذیری این سد حیاتی افزایش مییابد.
یکی از بینشهای عمیق در پاتوفیزیولوژی CAD، نقش نیروهای همودینامیک و مکانیکی خون بر رفتار سلولهای اندوتلیال است. پلاکهای آترواسکلروتیک به صورت تصادفی ایجاد نمیشوند؛ بلکه الگوی توزیع آنها قویاً با الگوهای جریان خون مرتبط است. نواحی مستقیم رگ که دارای جریان خون لایه ای (Laminar Flow) و تنش برشی بالا (High Shear Stress) هستند، در برابر آترواسکلروز مقاوماند. در این نواحی، تنش برشی باعث فعال شدن فاکتورهای رونویسی محافظتی مانند KLF2 و Nrf2 میشود که بیان ژن eNOS (تولیدکننده نیتریک اکساید) را افزایش داده و التهاب را سرکوب میکنند.
در مقابل، در نواحی دوچاخگیها (Bifurcations) و قوسهای شریانی، جریان خون آشفته (Disturbed Flow) و تنش برشی نوسانی (Oscillatory Shear Stress) حاکم است. این نیروی مکانیکی مخرب توسط گیرنده های مکانیکی سطح سلول حس شده و مسیرهای سیگنالینگ التهابی مانند NF-κB را فعال میکند. نتیجه این فرآیند، کاهش تولید NO، افزایش نفوذپذیری اندوتلیوم و بیان نابجای مولکولهای چسبنده (Adhesion Molecules) مانند VCAM-1 و ICAM-1 است که زمینه را برای نفوذ چربی و سلولهای ایمنی فراهم میکند. علاوه بر همودینامیک، عواملی مانند فشار خون بالا، هایپرگلیسمی و رادیکالهای آزاد ناشی از سیگار نیز مستقیماً به ساختار اندوتلیوم آسیب میرسانند.
۲.۲. نفوذ، به دام افتادن و اکسیداسیون لیپوپروتئینها
با نفوذپذیر شدن سد اندوتلیال، ذرات لیپوپروتئین با چگالی کم (LDL) وارد فضای زیرین اندوتلیوم (انتیما) میشوند. طبق «فرضیه پاسخ به احتباس» (Response-to-Retention Hypothesis)، ذرات LDL که وارد اینتیما میشوند، توسط پروتئوگلیکانهای ماتریکس خارج سلولی به دام میافتند. این به دام افتادن، زمان کافی را برای تغییرات شیمیایی خطرناک روی LDL فراهم میکند.
در محیط اکسیداتیو دیواره رگ، آنزیمهایی مانند میلوپراکسیداز (MPO) و لیپوکسیژنازها، به همراه رادیکالهای آزاد، باعث اکسیداسیون LDL میشوند و آن را به oxLDL تبدیل میکنند. مولکول oxLDL برای بدن یک موجود بیگانه و سمی تلقی میشود. این مولکول نه تنها باعث تحریک بیشتر التهاب میشود، بلکه مستقیماً به سلولهای اندوتلیال آسیب رسانده و حتی باعث مرگ سلولی آنها میشود. تحقیقات اخیر همچنین نقش مکانیسمهای اپیژنتیک مانند microRNAها و lncRNAها را در تنظیم این پاسخ التهابی و تغییر ماهیت سلولها در مواجهه با oxLDL برجسته کردهاند.
۲.۳. التهاب، فراخوان لکوسیتها و تشکیل سلولهای کفی
حضور oxLDL باعث میشود سلولهای اندوتلیال پیامهای خطر (S.O.S) صادر کنند. بیان مولکولهای چسبنده روی سطح اندوتلیوم باعث جذب مونوسیتهای در گردش خون میشود. این مونوسیتها به داخل دیواره رگ دیapedesis (مهاجرت) کرده و تحت تأثیر سیتوکینهایی مانند M-CSF به ماکروفاژ تبدیل میشوند.
[Image of atherosclerosis plaque formation]ماکروفاژها با هدف پاکسازی محیط، شروع به بلعیدن oxLDL میکنند. اما بر خلاف گیرنده LDL معمولی که دارای مکانیسم تنظیم بازخورد منفی است، گیرندههایی که مسئول برداشت oxLDL هستند (مانند گیرندههای Scavenger CD36 و LOX-1)، هیچ محدودیتی ندارند و تا زمانی که چربی وجود دارد، آن را میبلعند. این بلعیدن سیریناپذیر منجر به تجمع عظیم قطرات کلسترولی در سیتوپلاسم ماکروفاژها میشود و آنها را به سلولهای پر از چربی به نام «سلولهای کفی» (Foam Cells) تبدیل میکند. تجمع این سلولها اولین ضایعه قابل رویت آترواسکلروز یعنی «رگه چربی» (Fatty Streak) را ایجاد میکند.
نکته مهم در پاتوفیزیولوژی مدرن این است که التهاب در اینجا متوقف نمیشود. فعال شدن کمپلکس پروتئینی درون سلولی به نام «اینفلامازوم NLRP3» در پاسخ به کریستالهای کلسترول، باعث تولید سیتوکینهای قدرتمندی مانند IL-1β و IL-18 میشود که التهاب را در دیواره رگ شعلهور نگه میدارند.
۲.۴. تغییر فنوتیپ سلولهای عضلانی صاف (SMC Phenotype Switching)
در گذشته تصور میشد که سلولهای عضلانی صاف عروق (VSMCs) تنها نقش ساختمانی و انقباضی دارند. اما تحقیقات جدید نشان میدهد که این سلولها دارای پلاستیسیته (انعطافپذیری) بالایی هستند. در پاسخ به التهاب و فاکتورهای رشد، VSMCها از لایه مدیا به لایه اینتیما مهاجرت کرده و دچار «تغییر فنوتیپ» میشوند.
این سلولها ویژگی انقباضی خود را از دست داده و به حالت «سنتتیک» (Synthetic) در میآیند. در این حالت، آنها شروع به تکثیر و تولید ماتریکس خارج سلولی (مانند کلاژن و الاستین) میکنند که برای تشکیل «کلاهک فیبروزی» (Fibrous Cap) و پایدار نگه داشتن پلاک ضروری است. با این حال، این تغییر فنوتیپ شمشیری دو لبه است؛ زیرا برخی از این سلولها میتوانند به سلولهای شبهماکروفاژ تبدیل شده و بار التهابی پلاک را افزایش دهند، یا حتی به سلولهای شبهاستخوانی (Osteochondrogenic) تمایز یافته و باعث کلسیفیکاسیون (رسوب کلسیم) در پلاک شوند. مسیرهای سیگنالینگ Notch نقش حیاتی در تنظیم این تغییر ماهیت و تعیین سرنوشت سلولهای عضلانی صاف در پلاک ایفا میکنند.
۲.۵. تشکیل هسته نکروتیک و ناپایداری پلاک
با پیشرفت بیماری، سلولهای کفی دچار آپوپتوز (مرگ برنامهریزی شده) میشوند. در حالت طبیعی، ماکروفاژهای دیگر باید این سلولهای مرده را پاکسازی کنند (فرآیندی به نام Efferocytosis). اما در محیط سمی پلاک پیشرفته، این مکانیسم پاکسازی مختل میشود. در نتیجه، سلولهای مرده متلاشی شده و محتویات چربی، آنزیمهای مخرب و عوامل لختهزای خود را به فضای خارج سلولی میریزند.
تجمع این مواد منجر به تشکیل یک «هسته نکروتیک» (Necrotic Core) نرم، پرچرب و فاقد سلول در مرکز پلاک میشود. همزمان، ماکروفاژهای فعال با ترشح آنزیمهایی به نام متالوپروتئینازهای ماتریکس (MMPs)، کلاژنهای کلاهک فیبروزی را تجزیه میکنند. اگر تعادل به نفع تخریب کلاژن باشد، کلاهک فیبروزی نازک و نازکتر میشود تا جایی که پلاک تبدیل به یک «فیبروآتروم با کلاهک نازک» (Thin-Cap Fibroatheroma – TCFA) میگردد. این نوع پلاک، که اغلب دارای هسته نکروتیک بزرگ و کلاهک نازک (کمتر از ۶۵ میکرون) است، مستعدترین ضایعه برای پارگی ناگهانی و ایجاد حمله قلبی است.
۳. علل و عوامل خطر: شبکه درهمتنیده متابولیک و محیطی
عوامل خطر آترواسکلروز تنها متغیرهای آماری نیستند، بلکه هر کدام از طریق مسیرهای مولکولی مشخصی به فرآیند بیماری شتاب میدهند.
۳.۱. دیابت و مقاومت به انسولین: تخریب متابولیک عروق
دیابت نوع ۲ و مقاومت به انسولین از قویترین محرکهای CAD هستند. مکانیسمهای آسیبرسان در دیابت فراتر از هایپرگلیسمی ساده است. مقاومت به انسولین باعث ایجاد یک عدم تعادل حیاتی در سیگنالینگ سلولهای اندوتلیال میشود: مسیر PI3K (که مسئول تولید NO و بقای سلول است) مختل میشود، در حالی که مسیر MAPK (که مسئول التهاب، انقباض عروق و تکثیر سلولی است) دستنخورده باقی مانده و حتی بیشفعال میشود. این عدم تعادل منجر به اختلال عملکرد شدید اندوتلیال میشود. علاوه بر این، قند خون بالا باعث تشکیل «محصولات نهایی گلیکاسیون پیشرفته» (AGEs) میشود که با اتصال به گیرندههای RAGE، استرس اکسیداتیو و التهاب را تشدید میکنند.
۳.۲. سیگار: بمباران شیمیایی اندوتلیوم
دود سیگار حاوی هزاران ماده سمی و رادیکال آزاد است که مستقیماً به عروق آسیب میزنند. یکی از مکانیسمهای کلیدی، تخلیه کوفاکتوری به نام تتراهیدروبیوپترین (BH4) است. بدون BH4 کافی، آنزیم eNOS (که باید NO بسازد) «جفتنشده» (Uncoupled) میشود. در این حالت وحشتناک، آنزیم eNOS به جای تولید نیتریک اکساید محافظ، شروع به تولید سوپراکسید (یک رادیکال آزاد مخرب) میکند که چرخه معیوب استرس اکسیداتیو را تسریع میبخشد. همچنین سیگار باعث افزایش ویسکوزیته خون و فعالیت پلاکتها شده و محیط را برای تشکیل لخته آماده میکند.
۳.۳. لیپیدها و لیپوپروتئینها
علاوه بر LDL کلاسیک، نقش ذرات دیگر نیز برجسته شده است. لیپوپروتئین (a) یا Lp(a)، که سطح آن عمدتاً ژنتیکی است، حامل فسفولیپیدهای اکسید شده است و هم خاصیت آتروژنیک (پلاکساز) و هم ترومبوژنیک (لختهساز) دارد. همچنین تریگلیسیرید بالا و HDL پایین (Dyslipidemia) که اغلب در سندرم متابولیک دیده میشوند، با تولید ذرات LDL کوچک و متراکم (sdLDL) که نفوذپذیری بالاتری به دیواره رگ دارند، خطر را افزایش میدهند.
۴. علائم بالینی و تشخیص: از آناتومی تا عملکرد
تشخیص CAD از تمرکز صرف بر “تنگی مجرا” به سمت ارزیابی “بار پلاک” و “ترکیب پلاک” تغییر جهت داده است.
۴.۱. طیف علائم و مکانیسمها
- ایسکمی خاموش: به دلیل پدیده گلاگوف، بسیاری از بیماران علیرغم داشتن پلاکهای حجیم، بدون علامت هستند.
- آنژین پایدار: ناشی از عدم تعادل عرضه و تقاضای اکسیژن در هنگام فعالیت است، زمانی که تنگی ثابت مانع افزایش جریان خون میشود.
- سندرمهای حاد کرونری (ACS): شامل آنژین ناپایدار و انفارکتوس میوکارد (MI) است. پاتولوژی زمینه ACS معمولاً «پارگی پلاک» (Plaque Rupture) است که در آن کلاهک فیبروزی پاره شده و هسته نکروتیک با خون تماس مییابد. مکانیسم دیگر، «فرسایش پلاک» (Plaque Erosion) است که در آن اندوتلیوم سطحی کنده میشود، بدون اینکه کلاهک عمیقاً پاره شود؛ این حالت در زنان جوانتر و سیگاریها شایعتر است.
۴.۲. ابزارهای تشخیصی نوین و ویژگیهای پلاک پرخطر
| روش تشخیصی | تمرکز اصلی | ویژگیهای کلیدی قابل تشخیص | ارزش بالینی و مکانیکی |
|---|---|---|---|
| نمره کلسیم (CAC Score) | بار کلی آترواسکلروز | کلسیم متراکم (Dense) vs کلسیم نقطهای (Spotty) | نمره صفر احتمال خطر را بسیار کم میکند. کلسیم نقطهای و ریز با ناپایداری پلاک مرتبط است. |
| آنژیوگرافی سیتی (CCTA) | آناتومی دیواره و لومن | علامت حلقه دستمال سفره (Napkin-Ring Sign)، پلاک کمتراکم | این روش قادر است ویژگیهای پلاک آسیبپذیر را شناسایی کند. “علامت حلقه دستمال سفره” پیشبینیکننده قوی حوادث قلبی آینده است. |
| آنژیوگرافی تهاجمی | لومن رگ | تنگی مجرا (Stenosis) | استاندارد طلایی برای دیدن تنگی، اما ناتوان در دیدن دیواره رگ و پلاکهایی که به سمت بیرون رشد کردهاند (پدیده گلاگوف). |
۵. مدیریت و درمان: هدفگیری پاتوفیزیولوژی
استراتژیهای درمانی مدرن بر پایه متوقف کردن فرآیند آترواسکلروز و تثبیت پلاک استوار هستند.
۵.۱. اصلاح سبک زندگی: مکانیسم مولکولی ورزش
ورزش تنها کالریسوزی نیست؛ ورزش یک داروی قوی برای اندوتلیوم است. افزایش جریان خون در حین ورزش باعث افزایش تنش برشی لامینار میشود. این نیرو بیان آنزیمهای آنتیاکسیدان (مانند سوپراکسید دیسموتاز) را افزایش داده و دسترسی زیستی به نیتریک اکساید را بهبود میبخشد که مستقیماً با التهاب و اختلال عملکرد اندوتلیال مقابله میکند.
۵.۲. مداخلات دارویی پیشرفته
استاتینها و فراتر از چربی: استاتینها علاوه بر کاهش LDL، دارای خواص «پلئوتروپیک» (Pleiotropic) هستند. آنها با مهار ایزوپرنوئیدها، فعالیت پروتئینهای کوچکی مانند Rac1 را مهار میکنند که نتیجه آن کاهش استرس اکسیداتیو و التهاب در دیواره رگ است. استاتینها کلاهک فیبروزی را ضخیمتر کرده و پلاک را در برابر پارگی مقاوم میکنند.
مهارکنندههای PCSK9: این داروها (مانند Evolocumab و Alirocumab) با جلوگیری از تخریب گیرندههای LDL در کبد، سطح کلسترول خون را به شدت کاهش میدهند. مطالعات نشان دادهاند که این کاهش شدید میتواند منجر به «پسرفت پلاک» (Plaque Regression) شود.
درمان ضد التهابی هدفمند (کلشیسین): ظهور کلشیسین با دوز پایین به عنوان یک درمان برای CAD، تأییدی بر ماهیت التهابی این بیماری است. کلشیسین با مهار اختصاصی اینفلامازوم NLRP3 در نوتروفیلها و ماکروفاژها، از ترشح سیتوکینهای التهابی جلوگیری میکند و ریسک حوادث قلبی را کاهش میدهد.
۵.۳. تصمیمگیری برای بازسازی عروق: استنت یا جراحی؟
انتخاب بین مداخله از طریق پوست (PCI/Stent) و جراحی بایپس (CABG) بر اساس ویژگیهای آناتومیک و بیولوژیک بیمار است.
- استنت (PCI): برای باز کردن تنگیهای موضعی و درمان سکتههای حاد عالی است. اما استنت تنها یک نقطه را درمان میکند و تاثیری بر سیر بیماری در سایر نقاط رگ ندارد.
- جراحی بایپس (CABG): در بیماران با درگیریهای متعدد (Multivessel) و دیابت، CABG ارجحیت دارد. مکانیسم برتری آن این است که گرافت جراحی، یک مسیر خونرسانی کاملاً جدید ایجاد میکند که کل بخش میانی رگ (که اغلب حاوی پلاکهای متعدد و منتشر است) را دور میزند. این کار بیمار را در برابر پارگی پلاکهای آینده در آن ناحیه محافظت میکند.
۶. نتیجهگیری
بیماری عروق کرونر یک وضعیت پویا و پیچیده است که از برهمکنش عوامل متابولیک (چربی، قند)، نیروهای مکانیکی (جریان خون) و پاسخ ایمنی بدن (التهاب) ناشی میشود. تغییر دیدگاه از “لوله گرفتگی” به “التهاب دیواره عروق” دریچههای جدیدی را برای تشخیص و درمان گشوده است. امروزه میدانیم که هدف درمان تنها باز کردن رگ نیست، بلکه “خاموش کردن” التهاب پلاک و “تثبیت” کلاهک فیبروزی از طریق داروها و اصلاح سبک زندگی است تا از فاجعه پارگی پلاک و حمله قلبی پیشگیری شود.
بازبینی توسط متخصص
بازبین علمی این مقاله
آیا عوامل خطر CAD در خانواده شما ارثی است؟
بسیاری از عوامل زمینهساز آترواسکلروز، مانند اختلالات چربی خون و التهاب، ریشههای ژنتیکی دارند. درک استعداد ژنتیکی شما میتواند به اتخاذ استراتژیهای پیشگیرانه دقیقتر و مؤثرتر کمک کند.
دریافت مشاوره ژنتیک