پاتوژنز مولکولی، منشأ سلولی و چشمانداز اپیدمیولوژیک کارسینوم هپاتوسلولار
نقشه یادگیری این مقاله
۱. پادکست: برای درک کلی، ابتدا به پادکست گوش دهید.
۲. ویدیو: ویدیو آموزشی را برای یادگیری عمیق مشاهده کنید.
۳. مطالعه متن: در نهایت، متن مقاله را به عنوان منبع جامع مرور کنید.
مشاهده ویدیو در آپارات
پاتوژنز مولکولی، منشأ سلولی و چشمانداز اپیدمیولوژیک کارسینوم هپاتوسلولار
از هپاتوسیتهای ترانسفورمه تا ریزمحیط متابولیک و سیروتیک
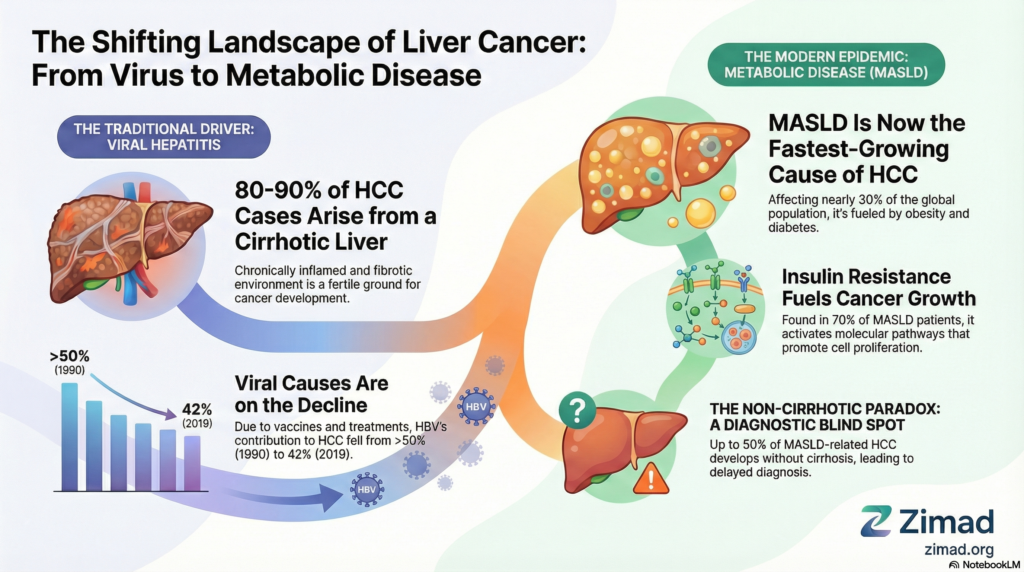
نئوپلاسم بدخیم اولیه کبد، که در اکثریت قریب به اتفاق موارد (۸۰ تا ۹۰ درصد) در قالب کارسینوم هپاتوسلولار (HCC) تظاهر مییابد، به عنوان یکی از مرگبارترین چالشهای حوزه انکولوژی در قرن بیست و یکم شناخته میشود. این بیماری در حال حاضر ششمین سرطان شایع و سومین علت اصلی مرگومیر ناشی از سرطان در سطح جهان است که بار اقتصادی و بهداشتی سنگینی را به ویژه در مناطق شرق آسیا و جنوب صحرای آفریقا تحمیل میکند. با وجود پیشرفتهای چشمگیر در روشهای تشخیصی، نرخ بقای پنجساله برای این بیماران همچنان در حدود ۱۸ درصد باقی مانده است که نشاندهنده ماهیت تهاجمی و پیچیدگیهای بیولوژیک این تومور است. در دهههای اخیر، دانش پزشکی شاهد یک تغییر پارادایم بنیادین در درک اتیولوژی و پاتوژنز این بیماری بوده است؛ جایی که محرکهای سنتی مانند عفونتهای ویروسی مزمن (HBV و HCV) جای خود را به محرکهای متابولیک ناشی از سبک زندگی مدرن، از جمله چاقی، دیابت و بیماری کبد استئاتوتیک مرتبط با اختلال عملکرد متابولیک (MASLD) میدهند. این گزارش به بررسی جامع منشأ سلولی HCC، نقش تعیینکننده ریزمحیط کبد سیروتیک و مکانیسمهای مولکولی زیربنایی در گذار از محرکهای ویروسی به متابولیک میپردازد.
تبیین منشأ سلولی: هپاتوسیت به عنوان سلول پایه در سرطانزایی کبد
شناسایی دقیق سلول منشأ (Cell of Origin) در کارسینوم هپاتوسلولار برای دههها موضوع بحثهای علمی گسترده بوده است. فرضیه سنتی بر این باور بود که سلولهای پیشساز کبدی (LPC) یا سلولهای بنیادی که در کانالهای هرینگ مستقر هستند، به دلیل توانایی نوسازی و پلاستیسیته بالا، کاندیدای اصلی ترانسفورماسیون بدخیم محسوب میشوند. با این حال، مطالعات نوین با بهرهگیری از تکنیکهای پیشرفته ردیابی دودمان (Lineage Tracing) در مدلهای حیوانی، شواهد متقاعدکنندهای ارائه دادهاند که نشان میدهد هپاتوسیتهای بالغ، و نه سلولهای پیشساز، منشأ اصلی HCC هستند.
شواهد تجربی از مدلهای ردیابی دودمان
در مدلهای سرطانزایی ژنوتوکسیک و ژنتیکی، استفاده از سیستمهای کروموزومی القایی مانند موشهای Opn-iCreERT2 Rosa26RYFP که به طور اختصاصی سلولهای پیشساز و مجاری صفراوی را برچسبگذاری میکنند، نشان داده است که هیچکدام از تومورهای HCC از این سلولها مشتق نشدهاند. در مقابل، زمانی که هپاتوسیتها با استفاده از ناقلهای AAV8-Tbg-Cre برچسبگذاری شدند، تمام تومورهای توسعهیافته در مدلهای موشی (از جمله مدل Mdr2KO که سیر پاتولوژیک التهاب، فیبروز و سرطان انسانی را بازسازی میکند) نشانگرهای فلورسنت هپاتوسیتی را بیان کردند. این یافتهها به طور قطع ثابت میکند که هپاتوسیتها سلولهای هدف برای جهشهای سرطانزا در کبد هستند.
تمایززدایی و پدیده سلولهای پیشساز کاذب
یکی از دلایل اصلی سردرگمی در شناسایی منشأ HCC، حضور نشانگرهای سلول پیشساز مانند سیتوکراتین ۱۹ (K19)، نشانگر A6 و آلفا-فیتوپروتئین (AFP) در بافت توموری است. تحقیقات نشان میدهند که بیان این نشانگرها نشاندهنده منشأ اولیه از سلولهای پیشساز نیست، بلکه بازتابی از تمایززدایی (Dedifferentiation) هپاتوسیتهای ترانسفورمه شده به یک فنوتیپ نابالغ و شبیه به سلولهای پروژنیتور است. این هپاتوسیتهای تمایززدوده شده که “امضای سلول پیشساز” را حمل میکنند، معمولاً با تومورهای تهاجمیتر و پیشآگهی بالینی بدتر همراه هستند.
| پارامتر مقایسهای | هپاتوسیتهای بالغ | سلولهای پیشساز کبدی (LPC) |
|---|---|---|
| فراوانی در پارانشیم کبد | > ۸۰٪ حجم کل کبد | بسیار نادر (< ۱٪ در کبد سالم) |
| نقش در بازسازی حاد | تکثیر مستقیم (Replication) | فعالسازی به عنوان ذخیره ثانویه |
| پتانسیل سرطانی در HCC | منشأ انحصاری تومورهای بدخیم | منشأ ضایعات خوشخیم و آدنومها |
| نشانگرهای بیولوژیک کلیدی | HNF4α, Albumin | K19, EpCAM, AFP |
| رفتار در طی تومورزایی | تمایززدایی به فنوتیپ نابالغ | فعالسازی پاراکرین توسط سلولهای سرطانی |
علاوه بر این، تعامل بین هپاتوسیتهای بدخیم و سلولهای پیشساز مجاور از اهمیت بالایی برخوردار است. هپاتوسیتهای ترانسفورمه شده از طریق ترشح سیگنالهای متابولیک و پاراکرین مانند گالکتین-۳ و آلفا-کتوگلوتارات (αKG)، باعث تحریک و گسترش سلولهای پیشساز در مراحل اولیه تومورزایی میشوند. این فرآیند به نوبه خود به ناهمگونی درونتوموری (Intra-tumoral Heterogeneity) کمک کرده و به سلولهای سرطانی اجازه میدهد تا در برابر فشارهای محیطی و درمانهای سیستمیک مقاومتر شوند.
ریزمحیط سیروتیک: از التهاب مزمن تا آشیانه پیشسرطانی
اکثریت قریب به اتفاق موارد کارسینوم هپاتوسلولار (تقریباً ۸۰ تا ۹۰ درصد) بر بستر سیروز کبدی ایجاد میشوند. سیروز که مرحله نهایی فیبروز پیشرفته کبد است، تنها یک تغییر ساختاری نیست، بلکه یک ریزمحیط (Microenvironment) بیوشیمیایی و سلولی بسیار پویا را فراهم میکند که به شدت مشوق ترانسفورماسیون نئوپلاستیک است. این ریزمحیط شامل تغییرات در ماتریکس خارج سلولی (ECM)، التهاب مداوم و اختلالات ایمنی سیستمیک است.
مکانیسمهای التهاب نکروماتوز و استرس اکسیداتیو
زنجیره وقایع از بیماری مزمن کبد به فیبروز و سپس سیروز، با التهاب نکروماتوز (Necroinflammation) مداوم آغاز میشود. در این محیط، مرگ مداوم هپاتوسیتها باعث آزاد شدن الگوهای مولکولی مرتبط با آسیب (DAMPs) میشود که به نوبه خود سلولهای ایمنی مقیم کبد مانند سلولهای کوپفر (KCs) و سلولهای غیرپارانشیمی مانند سلولهای ستارهای کبد (HSCs) را فعال میکند. فعال شدن مسیرهای سیگنالدهی کلیدی مانند NF-κB، STAT3 و JNK در این سلولها، پیوند گمشده بین التهاب مزمن و سرطان محسوب میشود. برای مثال، نشان داده شده است که فعالسازی دائمی NF-κB منجر به تولید کموکاینهایی مانند CXCL10، CXCL1 و CCL7 میشود که سلولهای ایمنی مختلفی را به کبد جذب کرده و یک حلقه بازخورد مثبت التهابی ایجاد میکنند.
نقش سلولهای غیرپارانشیمی در بازسازی ماتریکس
سلولهای ستارهای کبد (HSCs) پس از فعال شدن توسط سایتوکاینهای التهابی مانند TGF-β و PDGF، به مایوفیبروبلاستهای تولیدکننده کلاژن تبدیل میشوند. این فرآیند منجر به رسوب گسترده ماتریکس خارج سلولی و افزایش سفتی (Stiffness) بافت کبد میگردد. تغییرات فیزیکی و شیمیایی در ECM نه تنها سدی در برابر نفوذ داروهای شیمیدرمانی ایجاد میکند، بلکه از طریق فعالسازی گیرندههای مکانیکی در سطح هپاتوسیتها، مسیرهای پرولایفراتیو را تحریک مینماید. علاوه بر این، در کبد سیروتیک، عدم تعادل بین متالوپروتئینازهای ماتریکس (MMPs) و مهارکنندههای بافتی آنها (TIMPs) منجر به بازسازی دائمی اما ناقص بافت میشود که محیطی ایدهآل برای بقای کلونهای بدخیم است.
پیری سلولی و فنوتیپ ترشحی مرتبط با پیری (SASP)
یکی از جنبههای متمایز ریزمحیط سیروتیک، القای پیری سلولی (Senescence) در هپاتوسیتهای غیرتوموری به دلیل کوتاه شدن تلومرها و آسیبهای مداوم DNA است. سلولهای پیر، اگرچه توانایی تقسیم خود را از دست دادهاند، اما به شدت از نظر متابولیک فعال باقی میمانند و مجموعهای از فاکتورهای پیشسرطانی را ترشح میکنند که به “فنوتیپ ترشحی مرتبط با پیری” یا SASP معروف است. فاکتورهای SASP شامل سایتوکاینهای التهابی و فاکتورهای رشد هستند که میتوانند باعث تحریک تکثیر در سلولهای ترانسفورمه شده مجاور شوند که هنوز دچار پیری نشدهاند. این پدیده پارادوکسیکال نشان میدهد که چگونه پیری سلولی در کبد سیروتیک، به جای جلوگیری از سرطان، به تومورزایی کمک میکند.
سندرم اختلال عملکرد ایمنی مرتبط با سیروز (CAIDS)
پیشرفت به سمت سیروز با حالتی از نقص ایمنی پیچیده به نام “سندرم اختلال عملکرد ایمنی مرتبط با سیروز” (CAIDS) همراه است. CAIDS با دو ویژگی متضاد مشخص میشود: التهاب سیستمیک مزمن و فلج ایمنی موضعی. در این وضعیت، سلولهای کوپفر و سایر سلولهای ارائهدهنده آنتیژن دچار خستگی ایمنی (Immune Exhaustion) شده و قادر به شناسایی و حذف موثر سلولهای پیشسرطانی نیستند. علاوه بر این، افزایش جمعیت سلولهای T تنظیمکننده (Tregs) و سلولهای سرکوبگر مشتق از میلوئید (MDSC) در ریزمحیط کبد، باعث سرکوب پاسخهای لنفوسیتهای T سیتوتوکسیک (CD8+) میشود. این محیط “سرد” ایمنی (Cold Tumor Microenvironment) یکی از دلایل اصلی شکست ایمنیدرمانی در بسیاری از بیماران مبتلا به HCC سیروتیک است.
تحول اپیدمیولوژیک: گذار از محرکهای ویروسی به محرکهای متابولیک
در طول سه دهه گذشته، چشمانداز اتیولوژیک کارسینوم هپاتوسلولار در سطح جهانی دستخوش تغییری بیسابقه شده است. در حالی که عفونتهای ویروسی (HBV و HCV) به طور تاریخی مسئول اکثریت موارد HCC بودهاند، سهم آنها به دلیل برنامههای واکسیناسیون گسترده و درمانهای ضد ویروسی نوین در حال کاهش است. در مقابل، بیماریهای مرتبط با سبک زندگی و اختلالات متابولیک به سرعت در حال تبدیل شدن به محرکهای اصلی این بدخیمی هستند.
کاهش سهم ویروسها در سایه درمانهای نوین
بر اساس دادههای جهانی، سهم هپاتیت B در بروز HCC از بیش از ۵۰ درصد در سال ۱۹۹۰ به حدود ۴۲ درصد در سال ۲۰۱۹ کاهش یافته است. این کاهش در کشورهایی مانند کره جنوبی، تایوان و سنگاپور به دلیل واکسیناسیون همگانی نوزادان بسیار چشمگیر بوده است. به طور مشابه، معرفی داروهای ضد ویروسی با اثر مستقیم (DAAs) منجر به کاهش قابل توجه موارد HCC مرتبط با هپاتیت C در ژاپن و کشورهای اروپایی شده است. با این حال، به دلیل طولانی بودن دوره کمون از عفونت تا سرطان، بار بیماریهای ویروسی همچنان در میانمدت قابل توجه خواهد بود.
انفجار موارد مرتبط با MASLD و سندرم متابولیک
بیماری کبد استئاتوتیک مرتبط با اختلال عملکرد متابولیک (MASLD) – که پیشتر NAFLD نامیده میشد – اکنون به شایعترین بیماری مزمن کبد در جهان تبدیل شده است که حدود ۳۰ درصد از جمعیت بزرگسال کره زمین را تحت تأثیر قرار داده است. تداوم اپیدمی چاقی و دیابت نوع ۲ باعث شده است که MASLD به سریعترین علت رو به رشد HCC در کشورهای پردرآمد و حتی برخی کشورهای در حال توسعه تبدیل شود.
| متغیر آماری | موارد مرتبط با ویروس (HBV/HCV) | موارد مرتبط با متابولیک (MASLD) |
|---|---|---|
| روند زمانی (۱۹۹۰-۲۰۲۵) | کاهشی یا تثبیت شده | به شدت افزایشی |
| سن متوسط تشخیص | ۶۶ تا ۷۰ سال | ۷۳ سال و بالاتر |
| ارتباط با سیروز | > ۹۰٪ موارد | ۵۰٪ تا ۶۰٪ موارد |
| ویژگیهای همراه | بار ویروسی بالا، التهاب فعال | چاقی، دیابت، دیسلیپیدمی |
| چالش اصلی بالینی | مقاومت دارویی ویروس | غربالگری در کبد غیرسیروتیک |
پیشبینیهای مبتنی بر مدلسازی مارکوف نشان میدهند که بروز سالانه HCC مرتبط با MASLD در ایالات متحده تا سال ۲۰۳۰ به میزان ۱۳۷ درصد افزایش خواهد یافت. این موضوع نشاندهنده یک بحران بهداشت عمومی در حال ظهور است که نیازمند استراتژیهای پیشگیرانه جدید میباشد.
نقش الکل به عنوان یک عامل کمکی و مستقل
مصرف الکل همچنان یکی از ستونهای اصلی اتیولوژی HCC است، به ویژه در اروپا و آمریکای شمالی. با این حال، اهمیت الکل در تعامل آن با سایر عوامل خطر نهفته است. مصرف الکل نه تنها به طور مستقل باعث ایجاد استرس اکسیداتیو و آسیب DNA میشود، بلکه به طور همافزایی (Synergy) خطر ناشی از چاقی و دیابت را تشدید میکند. مطالعات کوهورت نشان دادهاند که در افراد چاق، حتی مصرف متوسط الکل میتواند خطر ابتلا به HCC را در مقایسه با افراد غیرچاق به شدت افزایش دهد.
پاتوژنز متابولیک: مکانیسمهای مولکولی در کبد استئاتوتیک
توسعه HCC در بستر MASLD از یک مسیر بیولوژیک متمایز پیروی میکند که در آن “ضربات موازی” (Parallel Hits) شامل مقاومت به انسولین، لیپوتوکسیسیته، استرس شبکه آندوپلاسمی و تغییرات در میکروبیوتای روده نقش دارند.
مقاومت به انسولین و محور IGF-1
مقاومت به انسولین (IR)، که در ۷۰ درصد بیماران MASLD مشاهده میشود، سنگ بنای پاتوژنز متابولیک است. هایپرانسولینمی ناشی از IR باعث تحریک تولید “فاکتور رشد شبهانسولین ۱” (IGF-1) در کبد میشود. هر دو هورمون انسولین و IGF-1 با اتصال به گیرندههای خود در سطح هپاتوسیتها، مسیرهای سیگنالدهی سرطانزا مانند PI3K/Akt/mTOR و MAPK/ERK را فعال میکنند که منجر به مهار آپوپتوز و تحریک تکثیر سلولی میشود. علاوه بر این، انسولین باعث مهار تولید پروتئینهای متصلشونده به IGF (IGFBPs) میشود که منجر به افزایش دسترسی بیولوژیک به IGF-1 در ریزمحیط کبد میگردد.
لیپوتوکسیسیته و استرس شبکه آندوپلاسمی (ER Stress)
تجمع بیش از حد لیپیدها در هپاتوسیتها منجر به تشکیل گونههای سمی چربی مانند سرامیدها و اسیدهای چرب اشباع (مانند اسید پالمیتیک) میشود. این بار اضافی لیپیدی باعث اختلال در تاخوردگی پروتئینها در شبکه آندوپلاسمی شده و وضعیتی به نام “استرس ER” ایجاد میکند. در پاسخ به این وضعیت، سلول مسیر “پاسخ پروتئینهای تا نشده” (UPR) را فعال میکند. اگرچه UPR در ابتدا تلاشی برای بازگرداندن هموستاز است، اما فعالسازی مزمن آن منجر به القای التهاب از طریق مسیرهای JNK و NF-κB و در نهایت مرگ سلولی یا ترانسفورماسیون بدخیم میشود.
استرس اکسیداتیو و آسیبهای میتوکندریایی
در MASLD، افزایش اکسیداسیون اسیدهای چرب در میتوکندریها و پراکسیزومها منجر به تولید بیش از حد گونههای فعال اکسیژن (ROS) و نیتروژن (RNS) میشود. این رادیکالهای آزاد باعث پراکسیداسیون لیپیدهای غشایی و آسیب مستقیم به DNA هپاتوسیتها میشوند. تجمع آسیبهای اکسیداتیو در DNA منجر به بروز جهشهای سوماتیک در ژنهای کلیدی مانند TERT (در ۶۰ درصد موارد)، TP53 (در ۳۰ درصد موارد) و CTNNB1 میشود. همچنین، اختلال در فرآیند میتوفاژی (حذف میتوکندریهای آسیبدیده) باعث تجمع میتوکندریهای ناکارآمد و تشدید تولید ROS میگردد که محیطی مستعد سرطان را فراهم میکند.
ارتباطات بینارگانی: محورهای چربی-کبد و روده-کبد
سرطان کبد ناشی از اختلالات متابولیک، پدیدهای منحصراً کبدی نیست، بلکه نتیجه یک اختلال سیستمیک در ارتباطات بینارگانی است که توسط وزیکولهای خارج سلولی (EVs)، آدیپوکاینها و محصولات میکروبی میانجیگری میشود.
محور بافت چربی-کبد: نقش آدیپوکاینها و اگزوزومها
بافت چربی در افراد چاق به عنوان یک ارگان غدد درونریز ناکارآمد عمل میکند. کاهش سطح آدیپونکتین (که دارای اثرات ضد التهابی و ضد سرطانی از طریق فعالسازی AMPK است) و افزایش سطح لپتین، باعث تحریک التهاب و فیبروز در کبد میشود. علاوه بر این، بافت چربی وزیکولهای خارج سلولی (اگزوزومها) حاوی microRNAهای خاصی را ترشح میکند که میتوانند مستقیماً متابولیسم کبد را تغییر دهند. برای مثال، اگزوزومهای حاوی miR-122 مشتق از بافت چربی باعث مهار پروتئین Sirt1 در هپاتوسیتها شده و از این طریق مسیر LKB1/AMPK را سرکوب و چربیزایی را تقویت میکنند.
محور روده-کبد و نقش دیسبیوزیس میکروبی
تغییر در ترکیب میکروبیوتای روده و افزایش نفوذپذیری روده (“روده نشتکننده”) در بیماران مبتلا به سندرم متابولیک، منجر به ورود مداوم لیپوپلیساکاریدها (LPS) و سایر الگوهای مولکولی مرتبط با میکروب (PAMPs) به جریان خون پورت میشود. این مواد با فعال کردن گیرندههای Toll-like (به ویژه TLR4) در سلولهای کوپفر و هپاتوسیتها، باعث ترشح سایتوکاینهای پیشالتهابی مانند TNF-α و IL-6 میشوند. همچنین، برخی متابولیتهای میکروبی مانند اسیدهای صفراوی ثانویه (مثلاً داکسیکولیک اسید) میتوانند با فعال کردن TLR2 در سلولهای ستارهای کبد، منجر به القای پیری در این سلولها و ترشح فاکتورهای SASP شوند که تومورزایی را تسریع میکند.
| محور ارتباطی | فاکتور میانجی کلیدی | اثر بر هپاتوسیتها | پیامد در تومورزایی |
|---|---|---|---|
| روده-کبد | لیپوپلیساکارید (LPS) | فعالسازی مسیر NF-κB | التهاب مزمن و القای جهش |
| چربی-کبد | اگزوزوم miR-22 | مهار بیان ژنهای متابولیک | القای اثر واربورگ و تکثیر |
| چربی-کبد | لپتین | تحریک مسیرهای بقا | مهار آپوپتوز سلولهای ترانسفورمه |
| روده-کبد | اسیدهای صفراوی ثانویه | فعالسازی TLR2 در HSCها | ترشح فاکتورهای رشد (SASP) |
پارادوکس کبد غیرسیروتیک در MASLD-HCC
یکی از چالشبرانگیزترین جنبههای بالینی HCC مرتبط با MASLD، وقوع آن در کبد غیرسیروتیک است. برخلاف هپاتیت C یا بیماری الکلی کبد که سرطان تقریباً همیشه در حضور سیروز رخ میدهد، در MASLD بین ۲۰ تا ۵۰ درصد موارد HCC در غیاب سیروز و حتی گاهی در غیاب فیبروز پیشرفته توسعه مییابند.
مکانیسمهای تومورزایی مستقل از فیبروز
در کبد غیرسیروتیک، مکانیسمهای سرطانزایی عمدتاً بر پایه استرس اکسیداتیو شدید و اختلالات سیگنالدهی انسولین استوار است. مطالعات بیوانفورماتیک نشان دادهاند که بیان ژنهای تنظیمکننده چرخه سلولی مانند CCNB1، E2F2 و CDK1 در موارد HCC غیرسیروتیک به طور قابلتوجهی بالاتر از موارد سیروتیک است. این موضوع نشان میدهد که در محیط کبد چرب، هپاتوسیتها حتی بدون نیاز به بازسازی ساختاری شدید بافت، پتانسیل بالایی برای عبور از نقاط بازرسی چرخه سلولی پیدا میکنند.
چالشهای غربالگری و پیامدهای بالینی
تومورهایی که در کبد غیرسیروتیک ایجاد میشوند، به دلیل نبود استراتژیهای غربالگری سیستماتیک برای بیماران MASLD غیرسیروتیک، معمولاً در مراحل بسیار پیشرفتهتر تشخیص داده میشوند. این تومورها اغلب بزرگتر هستند (میانگین ۶ سانتیمتر در مقابل ۴.۸ سانتیمتر در موارد ویروسی) و به دلیل عدم وجود سیروز، ممکن است به عنوان ضایعات خوشخیم تشخیص داده نشوند تا زمانی که علائم تهاجمی ظاهر شود. این موضوع منجر به یک تناقض بالینی شده است؛ جایی که بیماران با عملکرد کبد بهتر (بدون سیروز) به دلیل تشخیص دیرهنگام، نرخ بقای کمتری نسبت به بیماران سیروتیک تحت غربالگری دارند.
برنامهریزی مجدد متابولیک: اثر واربورگ در سلولهای توموری کبد
تطابق متابولیک، سنگ بنای بقای سلولهای بدخیم کبد در ریزمحیطهای چالشبرانگیز است. هپاتوسیتهای ترانسفورمه شده در بستر MASLD، متابولیسم خود را برای حمایت از تکثیر سریع بازسازی میکنند.
گلیکولیز هوازی و انحراف مسیرهای متابولیک
حتی در حضور اکسیژن کافی، سلولهای HCC ترجیح میدهند گلوکز را به لاکتات تبدیل کنند که به “اثر واربورگ” (Warburg Effect) معروف است. این فرآیند اگرچه از نظر تولید ATP بازده کمتری دارد، اما به سرعت واسطههای بیوسنتزی مورد نیاز برای سنتز نوکلئوتیدها، اسیدهای آمینه و لیپیدها را فراهم میکند. تغییرات اپیژنتیک و بیان microRNAهایی مانند miR-22-3p نقش کلیدی در مهار گلوکونئوژنز و القای گلیکولیز در هپاتوسیتهای سرطانی ایفا میکنند.
متابولیسم لیپید و گلوتامین
علاوه بر گلوکز، سلولهای سرطانی کبد به شدت به سنتز اسیدهای چرب “د نوو” (De novo Lipogenesis) وابسته هستند تا غشاهای سلولی جدید را بسازند. همزمان، اکسیداسیون اسیدهای چرب (β-oxidation) در این سلولها مهار میشود تا از ایجاد استرس اکسیداتیو بیش از حد جلوگیری شود. همچنین، مصرف بالای گلوتامین برای تامین نیتروژن مورد نیاز برای سنتز پورینها و پیریمیدینها و همچنین حفظ تعادل ردوکس از طریق تولید گلوتاتیون، یکی دیگر از ویژگیهای بارز متابولیک در HCC است.
رویکردهای نوین در پیشگیری و درمان
با توجه به تغییر ماهیت اتیولوژیک HCC، استراتژیهای درمانی در حال حرکت به سمت مداخلات متابولیک و ایمنی شخصیسازی شده هستند.
مداخلات سبک زندگی و جراحی باریاتریک
تغییر ساختار رژیم غذایی (رژیمهای کمکالری و کمکربوهیدرات) و فعالیت بدنی منظم (حداقل ۲ ساعت ورزش شدید در هفته) از طریق بهبود عملکرد میتوکندری و کاهش سایتوکاینهای التهابی، خطر ابتلا به HCC را به طور معنیداری کاهش میدهند. جالب توجه است که دادهها نشان میدهند جراحی باریاتریک در بیماران مبتلا به چاقی مفرط میتواند خطر پیشرفت MASLD به HCC را تا ۷۰ درصد کاهش دهد.
اهداف دارویی نوین: AMPK و SIRT7
مسیر AMPK: فعالسازی AMPK به عنوان یک مهارکننده طبیعی تومور عمل میکند که مسیرهای سنتز لیپید را سرکوب و حساسیت به انسولین را بهبود میبخشد. آگونیستهای AMPK و آنالوگهای FGF21 به عنوان کاندیداهای امیدوارکننده برای پیشگیری و درمان HCC مرتبط با متابولیک در حال بررسی هستند.
مسیر SIRT7: الکل و استرس اکسیداتیو باعث افزایش بیان SIRT7 میشوند که منجر به ارتقای متاستاز از طریق القای انتقال اپیتلیال به مزانشیمی (EMT) میگردد. مهار SIRT7 میتواند یک استراتژی درمانی هدفمند برای موارد تهاجمی HCC باشد.
چالشهای ایمونوتراپی و درمانهای ترکیبی
اگرچه ترکیب آتزولیزوماب و بواسیزوماب استاندارد طلایی درمان خط اول برای HCC پیشرفته است، شواهدی وجود دارد که نشان میدهد پاسخ به مهارکنندههای نقاط بازرسی ایمنی (ICIs) ممکن است در بیماران MASLD-HCC نسبت به بیماران ویروسی ضعیفتر باشد. این موضوع بر لزوم طراحی کارآزماییهای بالینی “اتیولوژی-محور” تاکید دارد که در آن وضعیت متابولیک بیمار به عنوان یک متغیر کلیدی در نظر گرفته شود.
نتیجهگیری و توصیههای راهبردی
کارسینوم هپاتوسلولار به عنوان یک نئوپلاسم بدخیم اولیه کبد، در حال تجربه یک تحول بنیادین است. هپاتوسیتهای بالغ به عنوان سلول منشأ، در معرض یک ریزمحیط به شدت سمی و التهابی قرار دارند که توسط سیروز یا اختلالات متابولیک تغذیه میشود. گذار از اتیولوژی ویروسی به محرکهای متابولیک مانند چاقی و الکل، نه تنها پاتوژنز بیماری را تغییر داده، بلکه چالشهای جدیدی را در تشخیص (وقوع سرطان در کبد غیرسیروتیک) و درمان ایجاد کرده است.
درک عمیق از محورهای ارتباطی بینارگانی و برنامهریزی مجدد متابولیک در سلولهای توموری، پنجرههای جدیدی را برای مداخلات درمانی باز کرده است. با این حال، برای کاهش بار جهانی این بیماری، تمرکز اصلی باید بر پیشگیری از سندرم متابولیک، بهینهسازی سیستمهای غربالگری فراتر از سیروز و تدوین پروتکلهای درمانی که هم تومور و هم بستر متابولیک آن را هدف قرار میدهند، معطوف گردد. آینده مدیریت HCC در گرو یک رویکرد چند رشتهای است که انکولوژی، غدد درونریز و هپاتولوژی را برای ارائه یک مراقبت شخصیسازی شده پیوند دهد.
بازبینی توسط متخصص
بازبین علمی این مقاله
آیا نگران سلامت کبد خود هستید؟
با توجه به افزایش شیوع کبد چرب و سرطان کبد، غربالگری ژنتیکی و متابولیک میتواند نجاتبخش باشد. اگر سابقه خانوادگی بیماری کبدی دارید، تیم ما آماده ارائه مشاوره تخصصی به شماست.
دریافت مشاوره ژنتیک