گزارش جامع پاتوبیولوژی و اپیدمیولوژی مولکولی سرطان کولورکتال
تحلیل توالیهای کارسینوژنز، تعاملات متابولیک میزبان و استراتژیهای مداخله
۱. مقدمه: چشمانداز بار جهانی و پارادایم بیماری
سرطان کولورکتال (Colorectal Cancer – CRC) به عنوان یکی از پیچیدهترین و در عین حال قابل پیشگیریترین بدخیمیهای دستگاه گوارش، نمادی از تعاملات چندلایه میان ژنتیک میزبان، تغییرات اپیژنتیک و عوامل محیطی است. این بیماری که سومین سرطان شایع و دومین علت مرگومیر ناشی از سرطان در سراسر جهان محسوب میشود، نه یک رویداد ناگهانی، بلکه نتیجهی یک فرآیند تکاملی چندمرحلهای است که طی آن اپیتلیوم طبیعی کولون از طریق انباشت تدریجی جهشهای سوماتیک و تغییرات مولکولی به یک توده بدخیم و تهاجمی تبدیل میشود. درک عمیق این فرآیند، که تحت عنوان “توالی آدنوماتوز-کارسینوم” شناخته میشود، سنگ بنای استراتژیهای مدرن در انکولوژی پیشگیرانه و درمانهای هدفمند است.
این گزارش با هدف ارائه یک تحلیل جامع و موشکافانه از پاتوژنز سرطان کولورکتال تدوین شده است. تمرکز اصلی بر ردیابی دقیق مسیرهای مولکولی است که یک سلول بنیادی در کریپتهای کولون را به سمت بدخیمی سوق میدهند. فراتر از ژنتیک، این گزارش به تحلیل نقش حیاتی عوامل سبک زندگی قابل اصلاح—بهویژه رژیم غذایی، چاقی و فعالیت بدنی—در تعدیل این مسیرهای مولکولی میپردازد. شواهد اپیدمیولوژیک و بیولوژیکی نشان میدهند که محیط متابولیک و التهابی بدن میزبان میتواند به عنوان یک کاتالیزور یا بازدارنده در فرآیند کارسینوژنز عمل کند. در نهایت، اهمیت حیاتی مکانیسمهای غربالگری به عنوان ابزاری برای قطع این زنجیره پاتولوژیک و جلوگیری از پیشرفت ضایعات پیشسرطانی به سرطان تهاجمی مورد بحث قرار خواهد گرفت.
آنچه سرطان کولورکتال را از بسیاری از نئوپلاسمهای دیگر متمایز میکند، وجود ضایعات پیشساز خوشخیم (پولیپها) است که پنجرهای زمانی طولانی—اغلب بیش از یک دهه—را برای مداخله فراهم میکنند. با این حال، ناهمگونی مولکولی این بیماری، که شامل مسیرهای کلاسیک ناپایداری کروموزومی (CIN) و مسیرهای متمایز دندانهدار (Serrated) و ناپایداری ریزماهوارهای (MSI) است، چالشهای قابل توجهی را در غربالگری و درمان ایجاد کرده است. این گزارش با بررسی دقیق متون علمی و دادههای پژوهشی، تلاش میکند تا تصویری یکپارچه از بیولوژی CRC ارائه دهد.
۲. پاتوبیولوژی مولکولی: کالبدشکافی توالی آدنوماتوز-کارسینوم
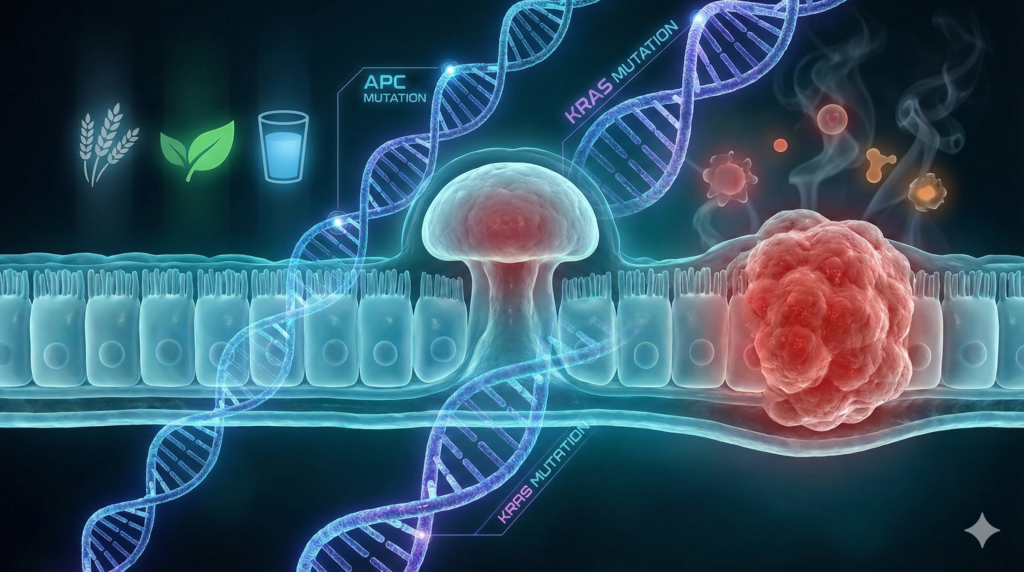
مدل توالی آدنوماتوز-کارسینوم که برای اولین بار توسط Fearon و Vogelstein در سال ۱۹۹۰ پیشنهاد شد، همچنان معتبرترین چارچوب برای درک اکثریت (حدود ۷۰ تا ۸۰ درصد) موارد سرطان کولورکتال اسپورادیک است. این مدل، سرطان را به عنوان یک بیماری ژنتیکی معرفی میکند که ناشی از انباشت متوالی و گامبهگام جهشهای سوماتیک در انکوژنها و ژنهای سرکوبگر تومور است. این مسیر عمدتاً با پدیدهای به نام ناپایداری کروموزومی (Chromosomal Instability – CIN) مشخص میشود که منجر به آنیوپلوئیدی (تعداد غیرطبیعی کروموزومها)، حذفها، تقویتها و جابجاییهای کروموزومی گسترده در سلولهای توموری میگردد.
۲.۱. آغازگر کارسینوژنز: اختلال در ژن APC و مسیر سیگنالینگ Wnt
رویداد آغازین و حیاتی در مسیر کلاسیک، که تقریباً در تمامی موارد با شروع تشکیل میکروآدنومها همراه است، غیرفعال شدن ژن APC (Adenomatous Polyposis Coli) واقع بر روی بازوی بلند کروموزوم ۵ (5q21) است. ژن APC به درستی به عنوان “دروازهبان” (Gatekeeper) کارسینوژنز کولورکتال شناخته میشود، زیرا عملکرد طبیعی آن مانع از ورود سلولهای اپیتلیال به فاز تکثیر کنترلنشده میشود.
مکانیسم مولکولی دقیق: کمپلکس تخریب و بتا-کاتنین
پروتئین APC نقش محوری در تنظیم مسیر سیگنالینگ Wnt دارد. در وضعیت هموستاز طبیعی کریپتهای کولون، APC به عنوان یک پروتئین داربستی (Scaffold protein) عمل میکند که اجزای “کمپلکس تخریب” (Destruction Complex) را کنار هم نگه میدارد. این کمپلکس شامل پروتئینهای Axin، گلیکوژن سنتاز کیناز ۳ بتا (GSK-3β) و کازئین کیناز ۱ (CK1) است.
- در غیاب جهش: کمپلکس تخریب به پروتئین سیتوپلاسمی بتا-کاتنین (β-catenin) متصل میشود. GSK-3β و CK1 با فسفوریله کردن بتا-کاتنین، آن را برای یوبیکوئیتیناسیون و تخریب سریع در پروتئازوم نشانهگذاری میکنند.
- در حضور جهش APC: پروتئین ناقص APC قادر به تشکیل کمپلکس تخریب کارآمد نیست. در نتیجه، بتا-کاتنین از تخریب فرار کرده و در سیتوپلاسم تجمع مییابد.
- انتقال به هسته و فعالسازی ژنها: بتا-کاتنین اضافی به هسته سلول منتقل میشود و با اتصال به فاکتورهای رونویسی خانواده TCF/LEF، بیان ژنهای هدف مسیر Wnt را روشن میکند.
پیامدهای فنوتیپی جهش APC: ژنهای هدف مسیر Wnt شامل تنظیمکنندگان کلیدی چرخه سلولی مانند c-MYC و Cyclin D1 هستند. نتیجه نهایی این اختلال، حفظ سلولهای اپیتلیال در وضعیت پرولیفراسیون مداوم (Stem-like state) و تشکیل کانونهای کریپت نابجا (ACF) و آدنومهای کوچک است.
۲.۲. شتابدهنده رشد: فعالسازی انکوژن KRAS
در حالی که جهش APC برای شروع آدنوم ضروری است، پیشرفت آن به یک آدنوم بزرگتر و دیسپلاستیکتر نیازمند رویدادهای ژنتیکی اضافی است. یکی از مهمترین این رویدادها، جهش فعالکننده در انکوژن KRAS است که در حدود ۴۰ تا ۵۰ درصد از آدنومهای بزرگ و کارسینومهای کولورکتال مشاهده میشود.
بیوشیمی KRAS و جهشهای کدون ۱۲ و ۱۳: ژن KRAS یک پروتئین GTPase کوچک را کد میکند. جهشهای KRAS در سرطان کولورکتال تقریباً منحصراً جهشهای نقطهای در کدونهای ۱۲ و ۱۳ هستند. این جهشها فعالیت GTPase ذاتی پروتئین را مختل میکنند و پروتئین KRAS را در حالت متصل به GTP و فعال “قفل” میکنند.
مسیرهای پاییندست و پیامدهای سلولی: فعالسازی دائمی KRAS مسیرهای RAF-MEK-ERK (MAPK) و PI3K-AKT-mTOR را تحریک میکند که منجر به گسترش کلونال سریع، افزایش اندازه پولیپ و تغییرات معماری مانند ویژگیهای ویلوس (Villous) میشود.
۲.۳. گذار به بدخیمی تهاجمی: غیرفعالسازی TP53 و ناپایداری کروموزومی
تبدیل نهایی یک آدنوم پیشرفته (با دیسپلازی درجه بالا) به کارسینوم تهاجمی معمولاً با غیرفعال شدن ژن سرکوبگر تومور TP53 مشخص میشود. این رویداد اغلب به عنوان “نقطه بیبازگشت” در مسیر کارسینوژنز در نظر گرفته میشود.
نقش نگهبان ژنوم: p53: پروتئین p53 به عنوان حسگر اصلی استرس سلولی عمل میکند و در پاسخ به آسیب DNA، توقف چرخه سلولی یا آپوپتوز را القا میکند. در سرطان کولورکتال، جهشهای TP53 باعث تولید پروتئین ناکارآمد میشوند. از دست دادن عملکرد p53 به سلولهای توموری اجازه میدهد تا با وجود آسیبهای ژنومی گسترده و آنیوپلوئیدی به تکثیر ادامه دهند و در برابر آپوپتوز مقاوم شوند.
۲.۴. نقش سایر مسیرها: TGF-β و SMAD4
حذف بازوی بلند کروموزوم ۱۸ (18q LOH) که حاوی ژن SMAD4 است، در حدود ۷۰ درصد سرطانهای کولورکتال رخ میدهد. غیرفعال شدن SMAD4 (واسط مسیر TGF-β) باعث میشود سلولهای توموری نسبت به اثرات مهارکننده رشد TGF-β مقاوم شوند.
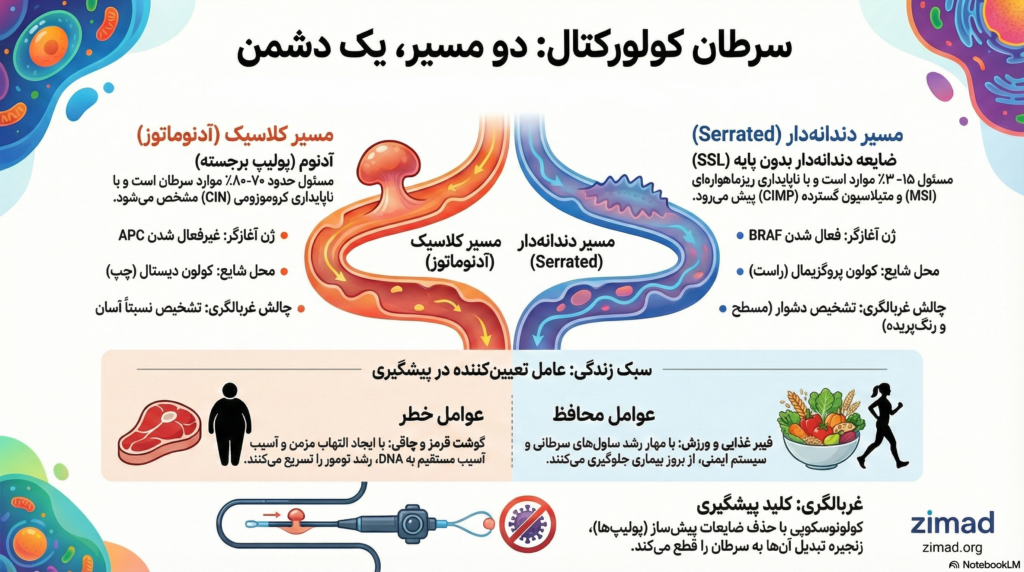
۳. مسیر نئوپلازی دندانهدار (Serrated Pathway): چالشی نوین در پاتولوژی و غربالگری
“مسیر دندانهدار” (Serrated Pathway) درک ما را از هتروژنیتی مولکولی CRC متحول کرده است. این مسیر مسئول ۱۵ تا ۳۰ درصد موارد سرطان کولورکتال است و از نظر بیولوژیکی و بالینی کاملاً متمایز است.
۳.۱. طبقهبندی هیستولوژیک و ضایعات پیشساز
برخلاف آدنومهای معمولی، ضایعات این مسیر دارای نمای “دندانارهای” در اپیتلیوم کریپتها هستند.
- پولیپهای هایپرپلاستیک (HPs): شایعترین نوع، اغلب کوچک و در کولون دیستال. پتانسیل بدخیمی پایینی دارند.
- ضایعات دندانهدار بدون پایه (SSLs): پیشسازهای اصلی سرطانهای مسیر دندانهدار. معمولاً بزرگتر، مسطح و در کولون پروگزیمال (راست) قرار دارند. تشخیص آنها در کولونوسکوپی بسیار دشوار است.
- آدنومهای دندانهدار سنتی (TSAs): کمتر شایع هستند و دارای ساختار بیرونزده میباشند.
۳.۲. مکانیسمهای مولکولی اختصاصی: مثلث BRAF، CIMP و MSI
مسیر دندانهدار با مجموعهای متمایز از رویدادهای مولکولی هدایت میشود:
- جهش آغازگر: BRAF V600E: برخلاف جهش APC، رویداد آغازین در اکثر SSLها، جهش فعالکننده در ژن BRAF است.
- فرار اپیژنتیک: فنوتیپ متیلاتور جزایر CpG (CIMP): برای غلبه بر سد پیری سلولی، ضایعات دندانهدار دچار متیلاسیون گسترده در پروموتر ژنهای سرکوبگر تومور میشوند.
- ناپایداری ریزماهوارهای (MSI): پیامد خاموشی MLH1: متیلاسیون پروموتر ژن MLH1 منجر به نقص در سیستم ترمیم عدم تطابق DNA (MMR) و وضعیت MSI-High میشود. این تومورها نرخ جهش بالایی دارند و کاندیدهای مناسبی برای ایمونوتراپی هستند.
| ویژگی | مسیر کلاسیک (آدنوماتوز) | مسیر دندانهدار (Serrated) |
|---|---|---|
| ژن آغازگر | APC (غیرفعالسازی) | BRAF (فعالسازی V600E) |
| مکانیسم اصلی | ناپایداری کروموزومی (CIN) | ناپایداری ریزماهوارهای (MSI) / CIMP |
| ضایعه پیشساز | آدنوم توبولار / ویلوس | پولیپ دندانهدار بدون پایه (SSL) |
| محل آناتومیک غالب | کولون دیستال (چپ) و رکتوم | کولون پروگزیمال (راست) |
| وضعیت متیلاسیون | پایین (CIMP-Low/Negative) | بالا (CIMP-High) |
| پروفایل ایمنی | “سرد” (Cold) – نفوذ ایمنی کم | “گرم” (Hot) – نفوذ لنفوسیتی بالا |
| چالش غربالگری | تشخیص نسبتاً آسان (برجستگی) | تشخیص دشوار (مسطح، رنگپریده) |
۴. تأثیر عوامل سبک زندگی بر پاتوژنز: مکانیسمهای متابولیک و مولکولی
سرطان کولورکتال یکی از حساسترین بدخیمیها نسبت به عوامل محیطی است. مطالعات مهاجرت نشان دادهاند که خطر ابتلا به این بیماری به شدت تحت تأثیر محیط است.
۴.۱. رژیم غذایی: شمشیر دو لبه کارسینوژنز
الف) گوشت قرمز و فرآوریشده: مصرف زیاد گوشت قرمز و فرآوریشده با افزایش خطر CRC مرتبط است. مکانیسمها شامل سمیت ژنی آهن هِم (تولید رادیکالهای آزاد)، تشکیل ترکیبات N-نیتروزو (عوامل آلکیلهکننده DNA) و آمینهای هتروسیکلیک (ناشی از پخت در دمای بالا) است.
ب) فیبر غذایی: فیبر غذایی اثر محافظتی دارد. فیبرهای محلول توسط باکتریهای روده تخمیر شده و بوتیرات تولید میکنند. بوتیرات به عنوان مهارکننده HDAC عمل کرده و بیان ژنهای سرکوبگر تومور را فعال میکند و باعث آپوپتوز سلولهای سرطانی میشود. فیبر نامحلول نیز با افزایش حجم مدفوع، غلظت کارسینوژنها را کاهش میدهد.
۴.۲. چاقی و بافت چربی: موتور محرک التهاب و رشد
چاقی، بهویژه چاقی شکمی، یکی از مهمترین عوامل خطر قابل اصلاح است.
مکانیسمهای ارتباط چاقی و سرطان:
- التهاب مزمن درجه پایین: بافت چربی سیتوکینهای التهابی (مانند IL-6) ترشح میکند که مسیرهای بقای سلولهای جهشیافته (STAT3) را فعال میکند.
- محور انسولین و IGF-1: هیپرانسولینمی و افزایش IGF-1 آزاد، مسیر PI3K/Akt/mTOR را فعال کرده و رشد سلولی را تحریک میکنند.
- عدم تعادل آدیپوکاینها: کاهش آدیپونکتین و افزایش لپتین، تهاجم سلولهای سرطانی را ترویج میکند.
پارادوکس چاقی: اگرچه چاقی خطر ابتلا را افزایش میدهد، برخی مطالعات نشان دادهاند که در بیماران مبتلا به مراحل پیشرفته، اضافه وزن ممکن است با بقای بهتر همراه باشد (احتمالاً به دلیل ذخایر انرژی بیشتر برای مقابله با کاشکسی).
۴.۳. فعالیت بدنی: تنظیمکننده سیستمیک
فعالیت بدنی منظم میتواند خطر CRC را ۲۰ تا ۲۵ درصد کاهش دهد. مکانیسمها شامل ترشح میوکینهای ضدتومور (مانند SPARC)، بهبود پروفایل متابولیک (کاهش انسولین) و افزایش حرکات روده است.
۴.۴. میکروبیوم روده: حلقه مفقوده
دیسبیوزیس و باکتریهای سرطانزا مانند Fusobacterium nucleatum در بافتهای تومورال CRC غنی میشوند. این باکتریها میتوانند مسیرهای سیگنالینگ سرطانزا را فعال کنند و التهاب سیستمیک را تشدید نمایند.
۵. اهمیت مکانیسمهای غربالگری و پایه بیولوژیکی تغییر بدخیم
با توجه به ماهیت مرحلهبهمرحلهی سرطانزایی در کولون، غربالگری مؤثرترین استراتژی برای کاهش بار این بیماری است. هدف نهایی غربالگری، قطع فیزیکی توالی آدنوماتوز-کارسینوم است.
۵.۱. پلیپکتومی: قطع زنجیره بیولوژیک
برداشتن آدنومها (پلیپکتومی) در حین کولونوسکوپی، توالی تکاملی سرطان را متوقف میکند. از آنجا که تبدیل یک آدنوم کوچک به کارسینوم تهاجمی ۱۰ تا ۱۵ سال طول میکشد، حذف پولیپ پتانسیل بدخیمی را کاملاً حذف میکند.
۵.۲. پایه بیولوژیکی تهاجم و متاستاز
زمانی که پیشگیری انجام نمیشود، آدنوم با کسب جهشهای اضافی (مانند TP53) به کارسینوم تبدیل میشود. این تغییر شامل تهاجم (توسط آنزیمهای MMP)، گذار اپیتلیال-مزانشیمال (EMT) و آنژیوژنز (رگزایی) است.
۵.۳. چالشهای غربالگری: بیولوژی سرطانهای فاصلهای
“سرطانهای فاصلهای” سرطانهایی هستند که در فاصله بین دو نوبت غربالگری ظاهر میشوند. اکثر این موارد ناشی از ضایعات دندانهدار بدون پایه (SSLs) هستند که در غربالگری اولیه نادیده گرفته شدهاند.
چرا SSLها نادیده گرفته میشوند؟ زیرا اغلب مسطح، رنگپریده و در کولون راست (جایی که آمادهسازی روده دشوارتر است) قرار دارند. به همین دلیل، افزایش کیفیت کولونوسکوپی و شناخت این ضایعات حیاتی است.
۵.۴. مقایسه روشهای غربالگری
- کولونوسکوپی: استاندارد طلایی، زیرا هم تشخیصی و هم درمانی است.
- تستهای ایمونوشیمیایی مدفوع (FIT): حساسیت بالا برای سرطان، اما کمتر برای آدنومهای پیشرفته.
- DNA مدفوع (Cologuard): ترکیب FIT با پنل مولکولی. حساسیت بالاتری برای تشخیص آدنومهای پیشرفته و ضایعات دندانهدار دارد.
۶. نتیجهگیری
سرطان کولورکتال بیماری است که از تقاطع ژنوم ناپایدار و محیط متابولیک التهابی پدید میآید. تحلیل مسیرهای مولکولی نشان میدهد که این بیماری یک موجودیت واحد نیست، بلکه مجموعهای از بیماریها است.
یافتههای این گزارش تأکید میکند که پیشگیری مولکولی از طریق اصلاح سبک زندگی امکانپذیر است، چاقی یک کارسینوژن فعال است و غربالگری باید هوشمندتر شود تا ضایعات مسیر دندانهدار را شناسایی کند. ادغام مداخلات سبک زندگی با برنامههای غربالگری دقیق، قدرتمندترین استراتژی برای کاهش بار این بیماری است.
| عامل سبک زندگی | اثر بر خطر CRC | مکانیسمهای بیولوژیکی و مولکولی اثبات شده |
|---|---|---|
| گوشت قرمز و فرآوریشده | افزایش خطر (↑) | تولید ترکیبات N-نیتروزو، کاتالیزوری ROS توسط آهن هِم، تشکیل آمینهای هتروسیکلیک. |
| فیبر غذایی | کاهش خطر (↓) | تولید بوتیرات (مهارکننده HDAC)، رقیقسازی اسیدهای صفراوی، کاهش زمان ترانزیت. |
| چاقی (بهویژه احشایی) | افزایش خطر (↑) | ترشح سیتوکینهای التهابی، هیپرانسولینمی و افزایش IGF-1 آزاد، تغییر نسبت لپتین/آدیپونکتین. |
| فعالیت بدنی | کاهش خطر (↓) | ترشح میوکینهای ضدتومور (SPARC)، فعالسازی NK cells، بهبود حساسیت به انسولین. |
| مصرف الکل | افزایش خطر (↑) | تولید استالدئید، تداخل با متابولیسم فولات، ایجاد استرس اکسیداتیو. |
بازبینی توسط متخصص
بازبین علمی این مقاله
آیا سابقه پولیپ یا سرطان روده در خانواده دارید؟
سرطان کولورکتال در بسیاری از موارد قابل پیشگیری است. اگر سابقه خانوادگی پولیپ روده یا سرطان دارید، مشاوره ژنتیک میتواند به تعیین سن شروع غربالگری و استراتژیهای پیشگیری کمک کند.
دریافت مشاوره ژنتیک