ژن چاقی یا ژن گرسنگی؟
نقشه یادگیری این مقاله
۱. پادکست: برای درک کلی، ابتدا به پادکست گوش دهید.
۲. ویدیو: ویدیو آموزشی را برای یادگیری عمیق مشاهده کنید.
۳. مطالعه متن: در نهایت، متن مقاله را به عنوان منبع جامع مرور کنید.
مشاهده ویدیو در آپارات
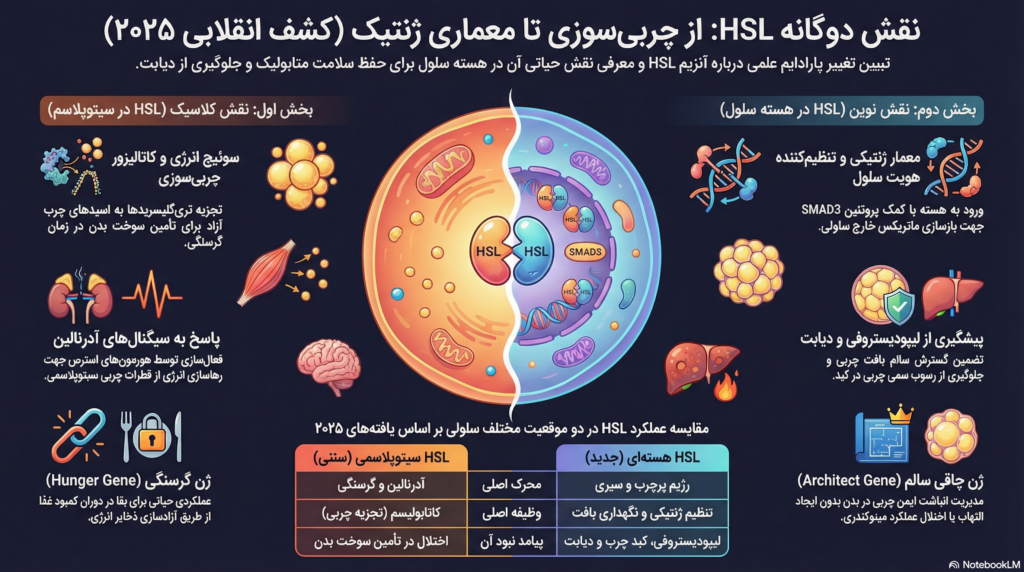
نقش دوگانه لیپاز حساس به هورمون (HSL) به عنوان تنظیمکننده ژنومیک هویت آدیپوسیت و هموستاز متابولیک
فراتر از کاتابولیسم چربی
مقدمه: بازنگری در یک پارادایم شصت ساله
برای بیش از شش دهه، لیپاز حساس به هورمون (HSL) به عنوان یکی از ارکان اصلی متابولیسم چربی شناخته میشد که نقش منحصر به فرد آن، کاتالیز کردن هیدرولیز ذخایر تریگلیسرید در بافت چربی سفید (WAT) برای تأمین سوخت ارگانهای جانبی در دوران کمبود انرژی بود. این آنزیم که در دهه ۱۹۶۰ کشف شد، همواره به عنوان یک “سوئیچ” انرژی توصیف شده است که تحت تأثیر محرکهای هورمونی مانند آدرنالین، اسیدهای چرب آزاد را از قطرات چربی (Lipid Droplets) رها میکند. با این حال، تحقیقات انقلابی منتشر شده در اکتبر ۲۰۲۵ در نشریه Cell Metabolism توسط تیم پروفسور دومینیک لانگن در دانشگاه تولوز، این دیدگاه کلاسیک را به طور بنیادین به چالش کشیده است.
یافتههای جدید نشان میدهند که پروتئین HSL فراتر از یک آنزیم سیتوپلاسمی ساده عمل میکند؛ این پروتئین قابلیت ورود به هسته سلولهای چربی (آدیپوسیتها) را دارد و در آنجا به عنوان یک فاکتور تنظیمکننده ژنتیکی، دستورات متابولیک سلول را تغییر میدهد. این کشف نه تنها معمای طولانیمدت “لیپودیستروفی” در بیماران فاقد HSL را حل میکند، بلکه مفهوم “ژن چاقی” و “ژن گرسنگی” را وارد ساحت جدیدی از درک بیولوژیک میسازد. در حالی که سیتوپلاسمی بودن HSL با پاسخ به گرسنگی مرتبط است، حضور هستهای آن برای حفظ هویت سلولی و گسترش سالم بافت چربی ضروری تشخیص داده شده است.
این گزارش به بررسی جامع سازوکارهای مولکولی انتقال HSL به هسته، تعامل آن با فاکتورهای رونویسی، تأثیر آن بر ماتریکس خارج سلولی و عملکرد میتوکندری، و پیامدهای بالینی این یافتهها در درمان دیابت و اختلالات متابولیک میپردازد.
بیوشیمی کلاسیک لیپوپروتئینها و مکانیسم سنتی HSL
پیش از پرداختن به نقش هستهای، درک عملکرد سنتی HSL در سیتوپلاسم برای تبیین تضاد عملکردی آن ضروری است. فرآیند لیپولیز یا تجزیه چربی در آدیپوسیتها یک توالی دقیق از واکنشهای آنزیمی است که توسط سیستم عصبی سمپاتیک و هورمونهای مختلف تنظیم میشود.
مسیر سیگنالدهی آدرنرژیک و فعالسازی HSL
در شرایط نیاز به انرژی، مانند روزهداری یا فعالیت بدنی، آدرنالین به گیرندههای بتای روی سطح آدیپوسیت متصل میشود و از طریق افزایش cAMP، پروتئین کیناز A (PKA) را فعال میکند. PKA سپس پروتئین HSL را در چندین باقیمانده سرین (مانند Ser563 و Ser659) فسفریله میکند. این فسفوریلاسیون باعث تغییر مکان HSL از سیتوزول به سطح قطرات چربی میشود، جایی که با پروتئینهایی مانند پریلیپین ۱ (PLIN1) تعامل میکند.
توالی آنزیمی تجزیه تریگلیسرید
لیپولیز توسط آنزیم ATGL (لیپاز تریگلیسرید چربی) آغاز میشود که تریگلیسرید را به دیگلیسرید تبدیل میکند. نقش HSL در این مرحله حیاتی است؛ زیرا این آنزیم مسئول اصلی تبدیل دیگلیسرید به مونوگلیسرید است، هرچند توانایی تجزیه تریگلیسرید و استرهای کلسترول را نیز دارد. بدون فعالیت سیتوپلاسمی HSL، جریان اسیدهای چرب به سمت عضلات و کبد مختل میشود.
| آنزیم | نقش اصلی در لیپولیز | محل فعالیت ترجیحی | وابستگی به PKA |
|---|---|---|---|
| ATGL | هیدرولیز تریگلیسرید به دیگلیسرید | سطح قطره چربی | کم (بیشتر وابسته به CGI-58) |
| HSL | هیدرولیز دیگلیسرید به مونوگلیسرید | سطح قطره چربی و هسته | بسیار زیاد (فسفوریلاسیون مستقیم) |
| MGL | هیدرولیز مونوگلیسرید به گلیسرول | سیتوپلاسم | ناچیز |
کشف ۲۰۲۵: HSL به عنوان یک فاکتور هستهای
تحقیقات سال ۲۰۲۵ نشان داد که HSL در هسته آدیپوسیتهای موش و انسان حضور دارد و این حضور تصادفی نیست، بلکه بخشی از یک برنامه تنظیمی دقیق برای حفظ توده بافت چربی است.
مکانیسم انتقال به هسته و تعامل با SMAD3
یکی از یافتههای کلیدی مطالعه “دوفو و همکاران” (Dufau et al.)، شناسایی تعامل پروتئین-پروتئین بین HSL و پروتئین SMAD3 است. SMAD3 یک پیامرسان کلیدی در مسیر سیگنالدهی TGF-β (فاکتور رشد تغییر شکلدهنده بتا) است که نقش مهمی در تمایز سلولی و پاسخهای فیبروتیک ایفا میکند. HSL با اتصال به SMAD3 وارد هسته شده و با کروماتین سلول پیوند میخورد. این انباشت هستهای به ویژه در شرایط تغذیه با رژیم غذایی پرچرب (HFD) افزایش مییابد که نشاندهنده نقش HSL در مدیریت گسترش بافت چربی است.
تنظیم متقابل توسط فسفوریلاسیون و صادرات هستهای
جالب است که فعالسازی HSL به عنوان یک لیپاز (توسط آدرنالین) اثر معکوسی بر حضور هستهای آن دارد. فسفوریلاسیون HSL توسط PKA که آن را به سمت قطرات چربی هدایت میکند، همزمان باعث خروج آن از هسته (Nuclear Export) میشود. این بدان معناست که در حالت روزهداری، سلول اولویت خود را از “تنظیم ژنتیکی و نگهداری بافت” به “تولید انرژی” تغییر میدهد. در مقابل، در حالت سیری یا چاقی، انباشت HSL در هسته به اوج خود میرسد تا برنامههای ژنتیکی مربوط به سلامت سلول را اجرا کند.
تغییر دستورات ژنتیکی: ماتریکس خارج سلولی و میتوکندری
نقش هستهای HSL مستقل از فعالیت کاتالیزوری (آنزیمی) آن است. این پروتئین در هسته دو برنامه ژنتیکی متضاد را مدیریت میکند که برای عملکرد صحیح آدیپوسیت حیاتی هستند.
ترویج بازسازی ماتریکس خارج سلولی (ECM)
بافت چربی برای گسترش سالم نیاز دارد که ماتریکس خارج سلولی خود را بازسازی کند تا فضای کافی برای آدیپوسیتهای بزرگ شده فراهم شود. HSL هستهای با تعامل با کمپلکسهای پروتئینی مانند SFPQ و NONO، رونویسی از ژنهای مرتبط با ECM را تقویت میکند. این فرآیند باعث میشود بافت چربی انعطافپذیر باقی بماند و دچار التهاب ناشی از هیپوکسی نشود.
سرکوب فسفوریلاسیون اکسیداتیو میتوکندری (OXPHOS)
در یک اقدام تنظیمکننده، HSL در هسته باعث کاهش بیان ژنهای مرتبط با فسفوریلاسیون اکسیداتیو در میتوکندری میشود. این سرکوب ممکن است به عنوان یک مکانیسم محافظتی برای جلوگیری از تولید بیش از حد گونههای اکسیژن فعال (ROS) در طول دورههای انباشت چربی عمل کند. با این حال، تعادل دقیق این فرآیند برای جلوگیری از اختلال عملکرد میتوکندری ضروری است، زیرا نقص در میتوکندریهای آدیپوسیت یکی از پیشزمینههای اصلی مقاومت به انسولین است.
| فرآیند سلولی | تأثیر HSL هستهای | پیامد بیولوژیک |
|---|---|---|
| بیوژنز ECM | افزایش رونویسی | گسترش سالم بافت و جلوگیری از فیبروز پاتولوژیک |
| عملکرد OXPHOS | کاهش رونویسی | مدیریت متابولیک و کنترل استرس اکسیداتیو |
| تعامل با SMAD3 | اتصال مستقیم | هماهنگی با مسیرهای تمایز سلولی TGF-β |
| پردازش RNA | پیوند با SFPQ/NONO | تنظیم پس از رونویسی دستورات متابولیک |
معمای لیپودیستروفی: چرا نبود HSL باعث لاغری پاتولوژیک میشود؟
یکی از پارادوکسهای بزرگ در بیولوژی بافت چربی این بود که چرا انسانها و موشهای دارای جهش در ژن HSL (ژن LIPE)، به جای چاق شدن (به دلیل عدم توانایی در تجزیه چربی)، دچار لیپودیستروفی یا کاهش شدید توده چربی میشوند. تحقیقات ۲۰۲۵ این معما را با تمرکز بر نقش هستهای HSL حل کرد.
شکست در برنامه نگهداری بافت
بدون HSL در هسته، آدیپوسیتها توانایی خود را برای حفظ ماتریکس خارج سلولی و هویت سلولی از دست میدهند. در نتیجه، بافت چربی نمیتواند به طور موثر منبسط شود. این پدیده منجر به یک وضعیت پاتولوژیک میشود که در آن چربیها به جای ذخیره شدن در بافت چربی زیرپوستی، در ارگانهای حیاتی مانند کبد و عضلات رسوب میکنند.
پیامدها: دیابت و استئاتوز کبدی
عدم گسترش سالم بافت چربی ناشی از نبود HSL، مستقیماً به مقاومت شدید به انسولین و دیابت نوع ۲ منجر میشود. تجمع نابجای چربی در کبد باعث بیماری کبد چرب متابولیک (MAFLD) میگردد. این یافتهها ثابت میکنند که چاقی لزوماً به معنای بافت چربی ناسالم نیست، بلکه ناتوانی در ذخیره چربی (لیپودیستروفی) به دلیل اختلال در نقش هستهای HSL میتواند به مراتب خطرناکتر باشد.
ژن چاقی یا ژن گرسنگی؟ بازنگری در اصطلاحات
عنوان “ژن چاقی یا ژن گرسنگی” نشاندهنده دوگانگی عملکردی HSL است که اکنون با کشف نقش هستهای آن معنای عمیقتری یافته است.
- HSL به عنوان ژن گرسنگی (سیتوپلاسم): در این نقش، HSL به عنوان یک کاتالیزور عمل میکند که در زمان نیاز بدن، ذخایر انرژی را آزاد میسازد. این عملکرد برای بقا در دوران کمبود غذا حیاتی است.
- HSL به عنوان ژن چاقی (هسته): در این نقش، HSL به عنوان یک معمار عمل میکند که اجازه میدهد بافت چربی به طور سالم رشد کرده و مقادیر زیادی انرژی را بدون آسیب به بدن ذخیره کند.
بنابراین، HSL نه تنها مسئول تجزیه چربی است، بلکه ضامن “سلامت” بافت چربی در دوران وفور نعمت نیز هست. نبود این ژن باعث میشود بدن حتی در حضور کالری زیاد، نتواند بافت چربی بسازد که منجر به فروپاشی متابولیک میگردد.
تعاملات میانبافتی و هموستاز سیستمیک
عملکرد HSL در هسته آدیپوسیت تنها به داخل سلول محدود نمیشود، بلکه بر کل شبکه متابولیک بدن تأثیر میگذارد. بافت چربی به عنوان یک ارگان اندوکرین، پیامهایی را به مغز، عضله و کبد ارسال میکند.
محور بافت چربی-عضله و نقش میوکینها
پروتئینهایی مانند ایریزین (Irisin) و METRNL که از عضلات ترشح میشوند، بر متابولیسم چربی و بیوژنز میتوکندری تأثیر میگذارند. برای مثال، ایریزین باعث “قهوهای شدن” بافت چربی سفید و افزایش ترموژنز میشود. تحقیقات نشان میدهد که این سیگنالهای خارجی ممکن است تعادل بین HSL هستهای و سیتوپلاسمی را تنظیم کنند تا پاسخهای متابولیک بدن به ورزش و سرما هماهنگ شود.
نقش میکروبیوتا در تنظیم HSL
شواهد جدید نشان میدهد که متابولیتهای میکروبی روده، مانند اسیدهای چرب کوتاه زنجیره (SCFAs)، از طریق مسیر TGF-β/SMAD3 بر اپیژنتیک و رسوب چربی در میزبان تأثیر میگذارند. از آنجایی که ورود HSL به هسته وابسته به SMAD3 است، وضعیت میکروبی روده میتواند به طور غیرمستقیم بر برنامه ژنتیکی آدیپوسیتها از طریق تغییر در سطح HSL هستهای تأثیر بگذارد.
| عامل تنظیمی | منشأ | تأثیر بر آدیپوسیت | ارتباط احتمالی با HSL |
|---|---|---|---|
| ایریزین | عضله | افزایش browning و UCP1 | تحریک خروج HSL از هسته برای اکسیداسیون |
| METRNL | عضله/قلب | ضد التهاب و بهبود حساسیت انسولین | تقویت مسیرهای بازسازی ECM |
| SCFAs | روده | تنظیم اپیژنتیک لیپیدها | تعدیل مسیر SMAD3 برای ورود HSL به هسته |
| آدرنالین | غدد فوق کلیوی | فعالسازی لیپولیز | صادرات HSL از هسته به سیتوپلاسم |
پتانسیلهای درمانی و چشمانداز آینده
کشف نقش دوگانه HSL افقهای جدیدی را در درمان بیماریهای متابولیک گشوده است. در حالی که پیش از این تمرکز بر مهار یا فعالسازی آنزیم HSL بود، اکنون استراتژیها به سمت تنظیم مکانمندی آن (Subcellular Localization) تغییر یافته است.
داروهای هدفمند برای جابجایی HSL
توسعه مولکولهایی که بتوانند ورود HSL به هسته را در بیماران مبتلا به لیپودیستروفی تسهیل کنند، یا خروج پاتولوژیک آن را در شرایط خاص مهار کنند، میتواند به بازگرداندن سلامت بافت چربی کمک کند. همچنین، در شرایط چاقی که HSL هستهای به طور غیرطبیعی افزایش مییابد، تعدیل این مسیر میتواند از فیبروز بافت چربی جلوگیری کند.
مدیریت دیابت نوع ۲
از آنجایی که نقص HSL هستهای مستقیماً با اختلال در گسترش بافت چربی و بروز دیابت در ارتباط است، هدف قرار دادن این پروتئین میتواند به عنوان یک درمان جایگزین یا مکمل برای داروهای فعلی مانند آگونیستهای GLP-1 (مانند تیرزپاتید) عمل کند. بازگرداندن “ظرفیت ذخیرهسازی سالم” به آدیپوسیتها از طریق HSL هستهای، میتواند بارهای سمی لیپید را از روی کبد و لوزالمعده بردارد.
نتیجهگیری: بازتعریف هویت آنزیم در قرن ۲۱
تحقیقات سال ۲۰۲۵ نشان داد که ما تنها نیمی از داستان HSL را میدانستیم. این پروتئین نه تنها “تخریبگر” چربی در سیتوپلاسم، بلکه “محافظ” هویت آدیپوسیت در هسته است. تغییر پارادایم از یک لیپاز ساده به یک تنظیمکننده ژنومیک، توضیح میدهد که چرا اختلال در این پروتئین به جای چاقی، به لیپودیستروفی و دیابت منجر میشود.
درک این نکته که HSL چگونه بین سیتوپلاسم و هسته در پاسخ به گرسنگی و سیری تعادل ایجاد میکند، کلید اصلی برای درمان بیماریهای متابولیک مدرن است. این آنزیم اکنون به عنوان یک پل ارتباطی بین متابولیسم لیپید و کنترل رونویسی شناخته میشود که هموستاز کل بدن را تضمین میکند. آینده تحقیقات در این حوزه احتمالاً بر شناسایی تمامی ژنهای تحت کنترل HSL و توسعه روشهای درمانی مبتنی بر جابجایی پروتئینی متمرکز خواهد بود.
بازبینی توسط متخصص
بازبین علمی این مقاله
آیا نگران اختلالات متابولیک و دیابت هستید؟
درک مکانیسمهای ژنتیکی و مولکولی مانند نقش HSL میتواند به تشخیص دقیقتر و درمانهای هدفمندتر برای دیابت و چاقی کمک کند. اگر سوالی در این زمینه دارید، تیم ما آماده ارائه مشاوره تخصصی است.
دریافت مشاوره ژنتیک