
اگزوم
نقشه یادگیری این مقاله
۱. پادکست: برای درک کلی، ابتدا به پادکست گوش دهید.
۲. ویدیو: ویدیو آموزشی را برای یادگیری عمیق مشاهده کنید.
۳. مطالعه متن: در نهایت، متن مقاله را به عنوان منبع جامع مرور کنید.
بزرگترین معما حل شد: رازی که در ۹۸٪ دیانای شما پنهان شده بود!
بازخوانی پارادایم ژنومیک و آغاز اودیسه تشخیصی در ماده تاریک ژنوم
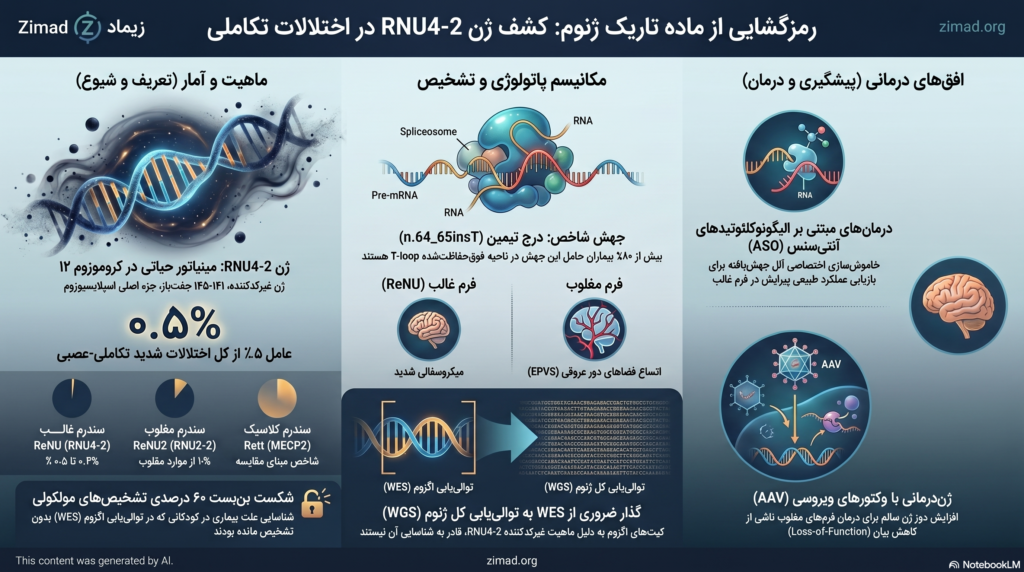
برای بیش از سه دهه، معماری ژنتیک پزشکی بر مبنای این فرضیه استوار بود که علل اصلی بیماریهای منوژنیک (تکژنی) شدید را باید در بخش کدکننده پروتئین ژنوم جستجو کرد. این ناحیه که «اگزوم» نام دارد، تنها حدود ۱.۵ درصد از کل ماده ژنتیکی انسان را تشکیل میدهد. با وجود پیشرفتهای چشمگیر در فناوری توالییابی کل اگزوم (WES)، نزدیک به ۶۰ درصد از کودکان مبتلا به اختلالات شدید تکاملی-عصبی (NDDs) پس از انجام آزمایشهای ژنتیکی استاندارد، همچنان بدون تشخیص مولکولی قطعی باقی میماندند. این بنبست بالینی طولانیمدت، فرضیهای انقلابی را در محافل آکادمیک تقویت کرد: پاسخ معما در اگزوم نیست، بلکه در پهنه وسیع ۹۸.۵ درصدی غیرکدکننده ژنوم، موسوم به «ماده تاریک ژنوم»، پنهان شده است.
در سال ۲۰۲۴، یک ائتلاف تحقیقاتی بینالمللی با آنالیز کلاندادههای ژنومیک موفق به کشف بزرگی شد که مسیر ژنتیک پزشکی را برای همیشه تغییر داد. دانشمندان دریافتند که جهشهای هتروزیگوت خودبهخودی (De novo) در یک ژن غیرکدکننده بسیار کوچک به نام RNU4-2، عامل اصلی هزاران مورد ناتوانی ذهنی و اختلالات رشدی تشخیصندادهشده در سراسر جهان است. این اکتشاف بیسابقه نشان داد که جامعه علمی بالینی برای سالها علت بیماریهای تکاملی را در جایگاههای اشتباهی جستجو میکرده است. اهمیت ژن RNU4-2 به حدی است که اکنون به عنوان یکی از شایعترین علل ژنتیکی تکژنی در پاتوژنولوژی اختلالات تکاملی-عصبی شناخته میشود و فراوانی آن با سندرمهای کلاسیک و شناختهشدهای مانند سندرم رت (Rett Syndrome) رقابت میکند.
معماری مولکولی ژن RNU4-2 و پویایی فیزیولوژیک آن در اسپلایسیوزوم
ژن RNU4-2 که روی بازوی بلند کروموزوم ۱۲ در موقعیت 12q24.23 قرار دارد، برخلاف ژنهای سنتی کدکننده پروتئین که اغلب دهها هزار جفتباز طول دارند، ژنی فوقالعاده مینیاتوری است و تنها ۱۴۱ الی ۱۴۵ جفتباز دارد. محصول رونویسی این ژن، یک RNA کوچک هستهای به نام U4 (U4 snRNA) است که به عنوان یکی از ستونهای ساختاری و تنظیمی اصلی در «اسپلایسیوزوم ماژور» عمل میکند. اسپلایسیوزوم یک ماشین ماکرومولکولی پویا و پیچیده است که وظیفه حذف دقیق اینترونها (بخشهای غیرکدکننده اولیه) و اتصال اگزونها (بخشهای کدکننده) را در پیشساز RNA پیامرسان (pre-mRNA) بر عهده دارد.
فرآیند کاتالیز اسپلایسیوزوم مستلزم تغییر آرایشهای ساختاری مکرر میان RNAهای کوچک هستهای است. در این پویایی مولکولی، U4 snRNA از طریق جفتشدن بازهای مکمل، یک کمپلکس دوپلکس فرعی با U6 snRNA تشکیل میدهد که نقش اصلی آن مهار فعالیت کاتالیتیکی پیشرس U6 تا زمان شناسایی دقیق مرز اینترون است. پس از ورود کمپلکس کاتالیتیکی به جایگاه واکنش، اسید نوکلئیک U4 از اَبَرکمپلکس باز شده و اجازه میدهد حلقه به شدت حساس و کاتالیتیکی U6 با جایگاه پیرایش ۵ پایانه (5′ splice site) جفت شود.
نقش تنظیمی برهمکنشهای U4/U6 به قدری حساس است که هرگونه اختلال در پایداری این دوپلکس، تعادل کل سیستم پیرایش سلولی را برهم میزند. ژن RNU4-2 در طول دوران تکوین جنینی مغز انسان به شدت فعال و دارای بیان بالایی است، امری که توجیه میکند چرا مغز در حال توسعه، نخستین و آسیبپذیرترین ارگان در برابر گسیختگی کاتالیتیکی اسپلایسیوزوم است.
مکانیسمهای پاتوفیزیولوژی مولکولی و طیف جهشهای پاتوژنیک
تحقیقات عمیق بیوشیمیایی و مدلسازیهای ساختاری سهبعدی با ابزارهای پیشرفتهای همچون AlphaFold3 و ChimeraX نشان میدهند که جهشهای پاتوژنیک در RNU4-2 ثبات فضایی دوپلکس U4/U6 را ویران میکنند. بیش از ۸۰ درصد از بیماران مبتلا به این بیماری هتروزیگوت، حامل یک جهش نقطهای مشخص به صورت درج یک جفتباز تیمین در موقعیتهای ۶۴ و ۶۵ ژنوم هستند که از نظر ژنتیکی به عنوان n.64_65insT شناخته میشود. از آنجا که ژن RNU4-2 روی رشته منفی (Reverse Strand) دیانای قرار دارد، این جهش درج تیمین در ژن، در دادههای توالییابی ژنومیک استاندارد سطح دیانای به صورت درج آدنین (T > TA در موقعیت کروموزومی GRCh38:chr12:120,291,839) گزارش میشود.
این درج تکنوکلئوتیدی در یک ناحیه هجده جفتبازی فوقالعاده حفاظتشده قرار دارد که لوپ-T (T-loop) و استم III (stem III) مولکول U4 را تشکیل میدهد. این ناحیه به طور فیزیکی موقعیت اسید نوکلئیکی U6 را برای شناسایی دقیق جایگاه پیرایش ۵ پایانه تعیین میکند. وقوع درج نوکلئوتیدی در لوپ-T پایداری این حلقه را از بین برده و مانع از گشودگی بهموقع دوپلکس U4/U6 میشود. این رخداد مکانیکی منجر به تغییر در انتخاب جایگاههای پیرایش ۵ پایانه طبیعی و ترجیح داده شدن جایگاههای پیرایش غیرطبیعی و فرعی (Cryptic Splice Sites) در هزاران ژن تکاملی مغز میشود.
علاوه بر جهش درج شاخص، واریانتهای نقطهای دیگری نیز شناسایی شدهاند که پیامدهای فنوتیپی متفاوتی دارند. برای نمونه، واریانت n.66A>G با تشنجهای مقاوم به درمان و رگرسیون شدید سیستم عصبی همراه است، در حالی که جهشهای مستقر در ناحیه استم III مانند n.76C>T فنوتیپهای به مراتب ملایمتری را با حفظ توانایی تکلم نسبی در بیماران ایجاد میکنند.
برای متخصصین ژنتیک پزشکی و آزمایشگاهی، شناسایی این لوکوس به دلیل شبهژنهای هومولوگ متعدد چالشبرانگیز است. از این رو، استفاده از پرایمرهای اختصاصی برای تکثیر واکنش زنجیرهای پلیمراز (PCR) و توالییابی مستقیم سنگر (Sanger Sequencing) در این ناحیه اهمیت بسزایی دارد. پرایمرهای بهینهسازیشده برای ردیابی این جهشها در جدول زیر ارایه شدهاند.
| پارامتر فنی ژنومیک | مشخصات ساختاری و آزمایشگاهی |
|---|---|
| موقعیت دقیق ژنومیک (hg38) | chr12:120,291,759 – 120,291,903 |
| شماره ترانسکرپت مرجع | NR_003137.3 |
| توالی پرایمر رفت (Forward Primer – 5′-> 3′) | GTT CCA ACA ACA AGA AAC CTC C |
| توالی پرایمر برگشت (Reverse Primer – 5′-> 3′) | TCA CGG AAT ACT CCT GAA CAA |
| طول قطعه تکثیرشده نهایی | تقریباً ۱۴۵ جفتباز |
پارادایم توارث دوگانه: ویرایش ژنوم اشباعکننده و کشف سندرم مغلوب RNU4-2
تا سال ۲۰۲۶، پنداشت کلی بر این بود که بیماریزایی ژن RNU4-2 صرفاً محدود به مکانیسم هتروزیگوت غالب است. با این حال، اعمال فناوری انقلابی ویرایش ژنوم اشباعکننده (Saturation Genome Editing – SGE) بر روی این لوکوس غیرکدکننده، این فرضیه را به طور کامل دگرگون ساخت. محققان با استفاده از سیستم CRISPR-Cas9، کتابخانهای از ۵۳۹ واریانت متمایز را در سراسر طول ژن RNU4-2 طراحی و وارد ردههای سلولی کردند تا اثر هر جهش را بر زیستپذیری سلولی بسنجند.
تحلیل آماری دادههای بقای سلولی و نمرات عملکردی حاصل از سنجش SGE منجر به کشف یک وراثت پنهان دیگر شد. دانشمندان متوجه شدند گروهی از جهشها که در نواحی بیرونی لوپ-T (مانند استم II، ساختار K-turn و جایگاه اتصال پروتئین Sm) قرار دارند، اثر ملایمتری بر بقای سلولی هتروزیگوت دارند، اما زمانی که به صورت هوموزیگوت یا هتروزیگوت مرکب (Biallelic) قرار میگیرند، فروریزش کاتالیتیکی شدیدی را ایجاد میکنند. با پیگیری این سرنخ در دیتابیسهای جهانی، ۳۸ بیمار مبتلا به اختلال تکاملی مغلوب ناشی از تغییرات دوآللی در RNU4-2 شناسایی شدند.
تفاوتهای بیولوژیکی و بالینی میان این دو الگوی توارثی بسیار چشمگیر است. در فرم غالب (سندرم ReNU)، علت بیماری اثر منفی غالب (Dominant-Negative) رونوشتهای لوپ-T بر روی پایداری کل اسپلایسیوزوم است. اما در فرم مغلوب جدید، پاتولوژی ناشی از کاهش شدید سطح رونوشتهای فعال ژن RNU4-2 (مکانیسم از دست رفتن عملکرد یا Loss-of-Function) است، پدیدهای که پایداری ساختار U4 را مختل کرده و منجر به افت شدید این RNA تنظیمی در سلولها میشود.
فنوتایپینگ بالینی و مقایسه آسیبشناسی ساختاری مغز در فرم غالب و مغلوب
بررسیهای بالینی دقیق بر روی بیماران مبتلا به اختلالات مرتبط با RNU4-2 نشان میدهد که هر دو فرم غالب و مغلوب با تاخیر جهانی در تکوین (GDD) و ناتوانی ذهنی (ID) تظاهر مییابند، اما جزئیات ساختاری مغز و ارگانهای دیگر تفاوتهای بنیادین دارند. در فرم مغلوب، اختلالات ساختاری خارج مغزی مانند کوتاهی مفرط قد یا ناهنجاریهای شدید اسکلتی کمتر دیده میشود، اما پاتولوژی ماده سفید مغز ویژگیهای بسیار متمایزی دارد.
تصویربرداریهای رزونانس مغناطیسی مغز (MRI) در بیماران فرم مغلوب فاش ساخت که بر خلاف فرم غالب که عمدتاً با کاهش عمومی حجم ماده سفید، آتروفی مخچه و اتساع بطنی همراه است، فرم مغلوب با اتساع شدید فضاهای دور عروقی (Enlarged Perivascular Spaces) در ماده سفید دور بطنی و عمیق مغز مشخص میشود. این فضاهای عروقی متسع در موارد پیشرفته فنوتیپی، الگویی شبیه به میکروسیستهای متراکم زنجیرهای را ایجاد میکنند که برای رادیولوژیستها به عنوان یک امضای پاتولوژیک تشخیصی عمل میکند. جزئیات مقایسهای فنوتیپهای بالینی در جدول زیر ترسیم شده است.
| مشخصه بالینی و پاتولوژیک | سندرم غالب ReNU (مونوآللی) | اختلال مغلوب RNU4-2 (دوباللی) |
|---|---|---|
| میزان شیوع تخمینی جهانی | حدود ۰.۴٪ الی ۰.۵٪ از بیماران NDD | حدود ۶۰٪ فراوانی فرم غالب در جوامع غیرخویشاوند |
| درجه ناتوانی ذهنی و رشدی | شدید تا عمیق (اغلب بیماران غیرکلامی هستند) | ملایم تا شدید (دارای توانایی گفتاری نسبی در برخی موارد) |
| میکروسفالی و کوتاهی قد | بسیار شایع (بالای ۷۰٪ بیماران) | ملایمتر یا غایب؛ عدم وجود کوتاهی مفرط قد |
| تشنج و اپیلپسی | شایع (حدود ۶۰٪) با پاسخ نسبی به دارو | وجود تشنج در مواردی خاص، فاقد الگوی صرع مقاوم پایدار |
| یافتههای MRI مغزی شاخص | هیپوپلازی جسم پینهای، ونتریکولومگالی، آتروفی مخچه | اتساع بارز فضاهای دور عروقی (الگوی میکروسیستی) |
| دیسمورفی چهره و اندامها | بسیار مشخص؛ چشمان گودرفته، تیغه بینی پهن، هیپوتونی شدید | خفیفتر؛ شباهتهای خانوادگی بیشتر از دیسمورفی عمومی |
بررسی اپیدمیولوژیک در کورتهای خویشاوندی و خانواده وسیع رنوپاتیها
یکی از دستاوردهای تشخیصی کلیدی در مطالعات اخیر، بررسی ژن RNU4-2 در جوامع با نرخ بالای ازدواجهای خویشاوندی (Consanguinity) بوده است. در پژوهشی جامع که توسط Bertoli-Avella و همکاران بر روی دادگان ژنومیک کمپانی Centogene (شامل ۲۲,۹۲۸ فرد توالییابی شده که ۴,۹۱۸ نفر از آنها مبتلا به اختلالات تکاملی-عصبی بودند) انجام گرفت، مشخص شد که علیرغم هتروزیکوت و غالب بودن واریانتهای پاتوژنیک ReNU، این بیماری بخش مهمی از بیماران جوامع خویشاوند را نیز به خود اختصاص میدهد. تقریباً ۵۰ درصد از بیماران مبتلا به NDD در این کورت دارای والدین خویشاوند بودند.
آزمایشها نشان دادند که واریانتهای تکژنی و هتروزیگوت RNU4-2 مسئول ۰.۵۵ درصد از موارد تاخیر رشدی در کل کورت و ۰.۲۵ درصد در زیرگروه خویشاوندان بودند. این یافته یک پیام بالینی بسیار حیاتی برای متخصصین ژنتیک دارد: در خانوادههای خویشاوند که همواره شک اولیه پزشک به سمت بیماریهای اتوزومال مغلوب میرود، نباید از احتمال جهشهای خودبهخودی و غالب در ژنهای غیرکدکننده نظیر RNU4-2 غافل شد.
علاوه بر RNU4-2، ژنوم انسان میزبان ژنهای اسپلایسیوزومی همخانواده دیگری نیز هست که به بروز رنوپاتیها (RNU-opathies) منجر میشوند. ژن RNU2-2 که پیشتر به عنوان یک شبهژن غیرفعال (RNU2-2P) طبقهبندی شده بود، اکنون به عنوان دومین عضو تاثیرگذار این خانواده شناخته میشود. ناهنجاریهای این لوکوس به دو فرم بالینی بروز میکنند: سندرم غالب ReNU2 (که حدود یکپنجم سندرم ReNU شیوع دارد) و سندرم مغلوب ReNU2 که توسط جهشهای دوباللی ایجاد میشود و با حاشیه امنیت فوقالعاده بالا، شایعترین علت اتوزومال مغلوب شناختهشده ناشی از یک ژن منفرد در ناتوانیهای رشدی انگلستان است.
| شاخص اپیدمیولوژیک | سندرم غالب ReNU (RNU4-2) | سندرم مغلوب ReNU2 (RNU2-2) | سندرم کلاسیک Rett (MECP2) |
|---|---|---|---|
| سهم از کل موارد NDD | ~ ۰.۴٪ الی ۰.۵٪ | ~ ۱۰٪ از کل موارد مغلوب | شاخص مبنا |
| شیوع تقریبی در جمعیت | ۱ در ۲۰,۰۰۰ الی ۳۰,۰۰۰ | بسیار بالا در جوامع هدف | ۱ در ۲۸,۰۰۰ (جنس مونث) |
| تعداد بیماران تقریبی جهانی | صدها هزار نفر | دهها هزار نفر | تخمین تثبیتشده بالینی |
| منبع دادههای اکتشاف | پروژه ۱۰۰ هزار ژنوم | پروژه ۱۰۰ هزار ژنوم | تاریخچه ژنتیک کلاسیک |
تحلیل علل ناکامی تاریخی بیوانفورماتیک بالینی در ردیابی RNU4-2
بررسی مسیر بیوانفورماتیکی پیش از سال ۲۰۲۴ نشان میدهد که ناتوانی در شناسایی جهشهای پاتوژنیک RNU4-2 تصادفی نبوده، بلکه ناشی از محدودیتهای سیستماتیک در سه لایه کلیدی بوده است:
- محدودیت ذاتی کپچر در اگزوم (WES): پروتکلهای غنیسازی اگزوم از کیتهای هیبریداسیونی استفاده میکنند که پروبهای آنها منحصراً برای هدفگیری اگزونهای پروتئینساز طراحی شدهاند. ژن RNU4-2 به عنوان یک ژن غیرکدکننده، عملاً در حین فرآیند غنیسازی حذف شده و پتانسیل بررسی پیدا نمیکرد.
- کوررنگی در خط لوله تراز کردن خوانشها (Read Alignment): به دلیل وجود شبهژنهای بیشمار اسپلایسیوزومی در سراسر ژنوم انسان که شباهت توالی بیش از ۹۵ درصدی با RNU4-2 دارند، ابزارهای نگاشت همردیفسازی (مانند BWA-MEM) خوانشهای کوتاه بهدستآمده از این ژن را به نواحی تکراری نامربوط نسبت میدادند.
- سوگیری در تحلیل اهمیت واریانتها (Variant Prioritization): در الگوریتمهای بالینی، همواره اولویت بررسی با جهشهای ایجادکننده کدون پایان یا تغییر فریم در پروتئینها بود. تغییرات تکباز در RNAهای غیرکدکننده به صورت سنتی فاقد پتانسیل تخریب بیولوژیکی تلقی میشدند.
عبور از این چالشها نیازمند مهاجرت به فناوری توالییابی کل ژنوم (WGS) و استفاده از پایپلاینهای اختصاصی همردیفسازی برای لوکوسهای غیرکدکننده فوقحفاظتشده بود.
افقهای درمانی نوین: پیوند مهندسی ژنتیک با مداخلات بالینی هدفمند
کشف نقش اساسی ژن RNU4-2 در گسیختگی کاتالیتیکی اسپلایسیوزوم، انقلابی را در استراتژیهای درمانی اختلالات تکاملی-عصبی آغاز کرده است. دو پلتفرم درمانی پیشرو در این حوزه در حال تکوین است:
- درمانهای مبتنی بر الیگونوکلئوتیدهای آنتیسنس (ASO): در سندرم غالب ReNU، هدف اصلی خاموشسازی اختصاصی آلل جهشیافته است. به کارگیری الیگونوکلئوتیدهای آنتیسنس مهندسیشده که به طور اختصاصی به توالی حاوی درج لوپ-T متصل میشوند، میتواند این ترانسکرپتهای سمی را برای تخریب توسط آنزیم RNase H نشانهگذاری کند.
- انتقال ژن مینیاتوری به کمک وکتورهای ویروسی (AAV-mediated Gene Therapy): در اختلالات مغلوب ناشی از کاهش بیان RNU4-2 و RNU2-2، استراتژی ایده آل، افزایش دوز ژن سالم است. کوچکی ابعاد این ژنها اجازه میدهد چندین نسخه فعال در یک وکتور ویروسی مرتبط با آدنو (AAV) بستهبندی و به سلولهای عصبی هدف منتقل شود.
نتیجهگیری و نقشه راه آینده ژنتیک بالینی
کشف نقش ژن غیرکدکننده RNU4-2 در پاتوژنولوژی اختلالات تکاملی-عصبی، پارادایمهای تشخیصی علم ژنتیک را برای همیشه تغییر داده است. این رخداد علمی نشان داد که بنبستهای تشخیصی در بخش بزرگی از بیماریهای ژنتیکی، نه به علت عدم وجود فاکتورهای منوژنیک، بلکه ناشی از محدودیت پهنای باند دید ما در اگزوم بوده است.
حل این معمای بزرگ، لزوم یک دگرگونی بنیادین در پلتفرمهای آزمایشگاهی را دیکته میکند: توالییابی کل ژنوم (WGS) دیگر نباید به عنوان یک تست لوکس و ثانویه، بلکه باید به عنوان خط اول غربالگریهای نوزادان و کودکان مبتلا به اختلالات رشدی در نظر گرفته شود. در حوزه درمان، سرعت بالای توسعه مدلهای سلولی و حیوانی با تکنیک SGE نویدبخش آن است که بیماران رنوپاتی نخستین گروه از کودکان مبتلا به ناتوانیهای ذهنی خواهند بود که درمانهای شخصیسازیشده و اصلاحکننده اسپلایسیوزوم را در سالهای پیش رو دریافت خواهند کرد.
بازبینی توسط متخصص
بازبین علمی این مقاله
آیا نگران تشخیص ناتوانی ذهنی فرزند خود هستید؟
کشف ژن RNU4-2 دریچهای نو به سوی تشخیص دقیق برای بیمارانی گشوده است که تستهای اگزوم آنها منفی بوده است. اگر فرزند شما دارای علائم تأخیر تکاملی بدون علت مشخص است، تیم متخصص زیماد آماده ارائه مشاوره ژنتیک و راهنمایی برای انجام تستهای نوین ژنومیک است.
دریافت مشاوره ژنتیک