
انقلاب در درمان کمخونی داسیشکل
تخریب معماری سهبعدی ژنوم و پارادایمهای نوین درمانی در گزارش جامع ۲۰۲۵ بیمارستان تحقیقاتی کودکان سنت جود
چکیده اجرایی
سال ۲۰۲۵ به عنوان نقطهی عطفی تاریخی در درمانهای ژنتیکی و مولکولی برای هموگلوبینوپاتیها، به ویژه بیماری کمخونی داسیشکل (Sickle Cell Disease – SCD) و تالاسمی بتا، ثبت شده است. گزارشهای منتشر شده از بیمارستان تحقیقاتی کودکان سنت جود (St. Jude Children’s Research Hospital) در این سال، نه تنها درک ما از مکانیسمهای دقیق ژندرمانیهای مبتنی بر CRISPR را دگرگون کرده است، بلکه دریچهای نوین به سوی درمانهای غیرویرایشی و دارویی گشوده است.
محور اصلی این تحول، کشف نقش حیاتی “معماری سهبعدی ژنوم” و ساختارهای “روزت کروماتینی” (Chromatin Rosette) در تنظیم بیان ژن BCL11A است. این گزارش جامع، با استناد به یافتههای منتشر شده در نوامبر ۲۰۲۵ در نشریه Blood و سایر پیشرفتهای بالینی و آزمایشگاهی سنت جود، به بررسی عمیق مکانیسمهای مولکولی، از جمله نقش RNAهای افزاینده (eRNAs) و پروتئین بارگذار کوهسین (NIPBL)، در حفظ ساختار سه بعدی DNA میپردازد. همچنین، پتانسیل استفاده از الیگونوکلئوتیدهای آنتیسنس (ASOs) به عنوان جایگزینی برای ویرایش ژنوم، نتایج کارآزماییهای بالینی در بهبود جریان خون مغزی و پیشگیری از سکته، و همچنین کشف اهداف ژنتیکی جدید مانند FLT1 مورد تحلیل قرار میگیرد. این سند با هدف ارائه تحلیلی تخصصی و جامع برای پژوهشگران و متخصصان حوزه هماتولوژی و ژنتیک مولکولی تدوین شده است.
۱. مقدمه: بستر پاتوفیزیولوژیک و ضرورت مداخله در سطح ژنوم
۱.۱ پاتولوژی مولکولی کمخونی داسیشکل و تالاسمی بتا
بیماری کمخونی داسیشکل، یکی از شایعترین و شدیدترین اختلالات تکژنی در جهان است که ناشی از یک جهش نقطهای (تغییر آدنین به تیمین) در ژن HBB میباشد. این ژن مسئول کدگذاری زیرواحد بتا-گلوبین در هموگلوبین بزرگسالان (HbA) است. جهش مذکور منجر به جایگزینی اسید آمینه گلوتامیک با والین در موقعیت ششم زنجیره بتا-گلوبین میشود و هموگلوبین داسی (HbS) را تولید میکند.
در شرایط کاهش اکسیژن (Deoxygenation)، تترادهای HbS پلیمریزه شده و رشتههای سخت و طویلی را تشکیل میدهند که باعث تغییر شکل گلبولهای قرمز به حالت داسیشکل (Sickle shape) میشوند. این سلولهای تغییرشکلیافته، انعطافپذیری خود را از دست داده و چسبندگی آنها به اندوتلیوم عروقی افزایش مییابد، که نتیجه آن انسداد عروق (Vaso-occlusion)، همولیز، کمخونی مزمن و آسیب پیشرونده به ارگانهای حیاتی است. به طور مشابه، تالاسمی بتا ناشی از جهشهایی است که تولید زنجیره بتا-گلوبین را کاهش داده یا متوقف میکنند، که منجر به اریتروپویزیس (خونسازی) غیرموثر و نیاز به تزریق مکرر خون میشود. هر دو بیماری ریشه در اختلال عملکرد هموگلوبین بزرگسالان دارند.
۱.۲ سوئیچ هموگلوبین جنینی به بزرگسال: هدف طلایی درمان
در انسان، بیان ژنهای گلوبین در طول توسعه تکاملی و رشدی تغییر میکند. در دوران جنینی، ژنهای گاما-گلوبین بیان میشوند و با آلفا-گلوبین ترکیب شده تا هموگلوبین جنینی (HbF) را بسازند. HbF تمایل بالایی به اکسیژن دارد و فاقد زیرواحد بتا است، بنابراین تحت تأثیر جهش داسیشکل قرار نمیگیرد. مدت کوتاهی پس از تولد، یک “سوئیچ رونویسی” (Transcriptional Switch) رخ میدهد: بیان گاما-گلوبین خاموش شده و بیان بتا-گلوبین فعال میشود.
مشاهدات بالینی نشان دادهاند که بیمارانی که به دلیل جهشهای طبیعی دیگر (مانند پایداری ارثی هموگلوبین جنینی یا HPFH)، سطوح بالای HbF را در بزرگسالی حفظ میکنند، علائم بسیار خفیفتری از بیماری داسیشکل را تجربه میکنند. HbF نه تنها جایگزین HbS میشود، بلکه به طور فعال از پلیمریزاسیون HbS جلوگیری میکند. از این رو، “فعالسازی مجدد هموگلوبین جنینی” به استراتژی اصلی درمانهای ژنتیکی مدرن تبدیل شده است.
۱.۳ نقش محوری BCL11A به عنوان سرکوبگر اصلی
از طریق مطالعات ارتباطی ژنومگستر (GWAS)، ژن BCL11A به عنوان تنظیمکننده کلیدی و سرکوبگر اصلی گاما-گلوبین شناسایی شد. پروتئین BCL11A با اتصال به نواحی تنظیمی در لوکوس بتا-گلوبین و فراخوانی کمپلکسهای سرکوبگر (مانند NuRD)، مانع از بیان ژنهای جنینی در سلولهای اریتروئید بالغ میشود.
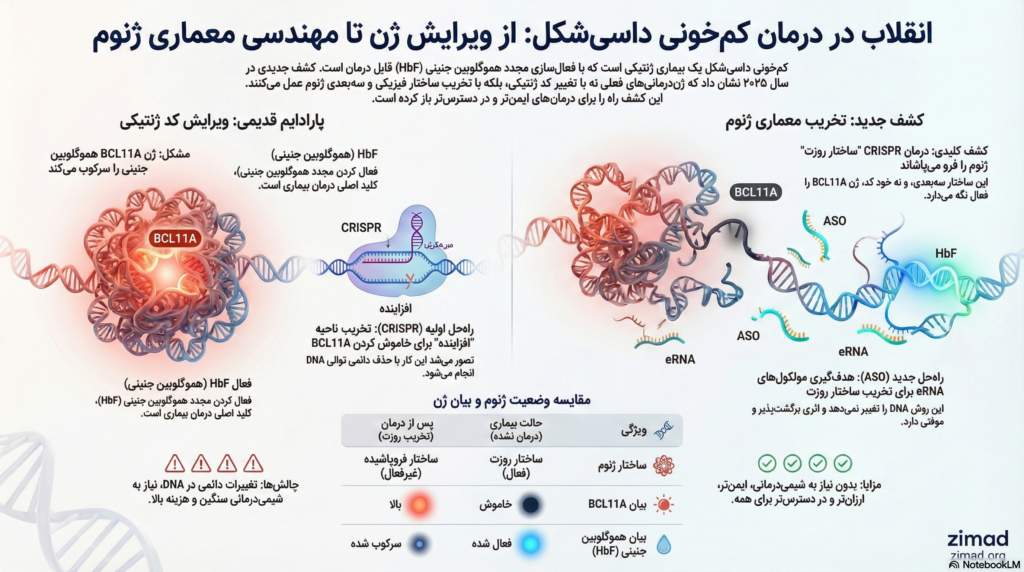
درمانهای ژنتیکی تایید شده اخیر، مانند Casgevy، از سیستم CRISPR-Cas9 برای ایجاد شکستهای دو رشتهای در ناحیه “افزاینده” (Enhancer) ژن BCL11A استفاده میکنند. این افزاینده که در فاصله ۵۸ کیلوبازی از نقطه شروع رونویسی قرار دارد، مختص سلولهای اریتروئید است. تخریب این ناحیه باعث کاهش بیان BCL11A و در نتیجه فعالسازی مجدد HbF میشود. تا پیش از گزارشهای سال ۲۰۲۵ سنت جود، تصور غالب بر این بود که تخریب توالی DNA مکانیسم اصلی است، اما یافتههای جدید نشان میدهد که داستان مبتنی بر ساختار فیزیکی ژنوم است.
۲. کشف انقلابی ۲۰۲۵: معماری کروماتین و ساختار “روزت”
در نوامبر ۲۰۲۵، تیم تحقیقاتی سنت جود مطالعهای جریانساز را منتشر کردند که مکانیسم مولکولی دقیق درمانهای CRISPR را بازتعریف کرد.
۲.۱ تعریف ساختار “روزت کروماتینی” (Chromatin Rosette)
با استفاده از تکنیکهای پیشرفته ضبط کانفورماسیون کروماتین، محققان دریافتند که افزاینده BCL11A در سلولهای پیشساز گلبول قرمز، یک ساختار خطی ساده نیست. بلکه این افزاینده به صورت فیزیکی تا خورده و یک ساختار سهبعدی پیچیده و گلمانند به نام “روزت کروماتینی” تشکیل میدهد.
این ساختار حاصل تعاملات فیزیکی متعدد بین افزاینده، پروموتر ژن BCL11A و سایر عناصر تنظیمی است. ساختار روزت به عنوان یک “عایق اپیژنتیکی” عمل میکند:
- تسهیل رونویسی: این ساختار با نزدیک کردن فیزیکی افزاینده و پروموتر، ماشین رونویسی را متمرکز کرده و بیان بالای BCL11A را تضمین میکند.
- حفاظت اپیژنتیکی: ساختار روزت مانع از ورود سیگنالهای سرکوبگر از نواحی مجاور به داخل ژن BCL11A میشود.
۲.۲ تخریب ساختار سه بعدی توسط CRISPR
یافته کلیدی گزارش ۲۰۲۵ این است که وقتی CRISPR-Cas9 در ناحیه افزاینده برش ایجاد میکند، اثر درمانی ناشی از حذف توالی نیست، بلکه ناشی از “فروپاشی ساختار روزت” است.
این مطالعه نشان داد که شکست DNA در این ناحیه باعث میشود که ساختار پیچیده سهبعدی از هم بپاشد. با از بین رفتن روزت:
- ارتباط فیزیکی بین افزاینده و پروموتر قطع میشود.
- عایق اپیژنتیکی از بین میرود.
- پروتئینهای سرکوبگر و نشانگرهای خاموشی به ناحیه هجوم آورده و ژن BCL11A را برای همیشه خاموش میکنند.
این کشف یک تغییر پارادایم بنیادی است: هدف درمان “تغییر کد ژنتیکی” نیست، بلکه “تخریب معماری فیزیکی” است که ژن بیماریزا را فعال نگه میدارد.
۳. مکانیسم مولکولی دقیق: نقش RNAهای افزاینده و NIPBL
برای درک اینکه چرا ساختار روزت فرو میریزد، محققان سنت جود دو بازیگر اصلی را شناسایی کردند: RNAهای افزاینده (eRNAs) و پروتئین NIPBL.
۳.۱ بیوژنز و عملکرد eRNA در لوکوس BCL11A
افزایندهها تنها توالیهای تنظیمکننده غیرفعال نیستند؛ آنها به طور فعال رونویسی میشوند و مولکولهای RNA غیرکدکنندهای به نام eRNA تولید میکنند. مطالعه ۲۰۲۵ ثابت کرد که eRNAهای تولید شده از افزاینده BCL11A نقشی ساختاری و حیاتی دارند. این eRNAها برای حفظ ساختار روزت ضروری هستند و احتمالاً به عنوان یک داربست عمل میکنند که پروتئینهای تشکیلدهنده حلقه کروماتین را در کنار هم نگه میدارند.
۳.۲ محور NIPBL-Cohesin و اکستروژن حلقه (Loop Extrusion)
یکی از بینشهای عمیق این پژوهش، ارتباط بین eRNA و بارگذاری کمپلکس کوهسین (Cohesin) است. کوهسین مسئول تشکیل حلقههای کروماتینی است و پروتئین NIPBL فاکتور اصلی بارگذارنده کوهسین بر روی DNA است.
زنجیره وقایع کشف شده توسط سنت جود:
- افزاینده BCL11A رونویسی شده و eRNA تولید میکند.
- eRNAها باعث جذب یا تثبیت NIPBL در ناحیه افزاینده میشوند.
- NIPBL کمپلکس کوهسین را روی کروماتین بارگذاری میکند.
- کوهسین با تشکیل حلقه، پروموتر و افزاینده را در ساختار روزت کنار هم قفل میکند.
- این ساختار باعث بیان بالای BCL11A و سرکوب HbF میشود.
زمانی که CRISPR یا هر عامل دیگری تولید eRNA را مختل کند، NIPBL جذب نمیشود، کوهسین بارگذاری نمیگردد و ساختار روزت فرو میریزد.
| ویژگی | وضعیت بیماری (درمان نشده) | وضعیت پس از درمان (تخریب روزت) |
|---|---|---|
| ساختار ژنوم | تشکیل روزت کروماتینی | فروپاشی ساختار سه بعدی و خطی شدن |
| وضعیت eRNA | بیان بالا و فعال | تخریب شده یا عدم بیان |
| اتصال NIPBL/Cohesin | بارگذاری فعال و پایدار | عدم بارگذاری و جدا شدن از کروماتین |
| وضعیت اپیژنتیکی | عایقبندی شده (فعال) | هجوم هتروکروماتین (خاموش) |
| بیان BCL11A | بالا (High) | خاموش (Silenced) |
| بیان هموگلوبین جنینی | سرکوب شده (Low) | فعال شده (Reactivated) |
۴. افق نوین درمانی: الیگونوکلئوتیدهای آنتیسنس (ASOs)
شاید مهمترین پیامد بالینی کشف مکانیسم eRNA، امکان استفاده از الیگونوکلئوتیدهای آنتیسنس (ASO) به جای ویرایش ژنوم باشد.
۴.۱ مکانیسم اثر ASO در برابر BCL11A
ASOها رشتههای کوتاه و مصنوعی هستند که میتوانند به RNA هدف متصل شوند و باعث تخریب آن شوند. محققان سنت جود ASOهایی طراحی کردند که به طور اختصاصی به eRNAهای افزاینده BCL11A متصل میشوند.
- نتایج آزمایشگاهی: تزریق این ASOها باعث تخریب سریع eRNAها شد.
- پیامد ساختاری: با حذف eRNA، دقیقاً مشابه اثر CRISPR، بارگذاری NIPBL مختل شد و ساختار روزت فرو ریخت.
- پیامد درمانی: بیان BCL11A خاموش شد و تولید هموگلوبین جنینی به سطوح درمانی افزایش یافت.
۴.۲ مزایا نسبت به ژندرمانی ویرایشی (CRISPR)
این رویکرد پتانسیل حل بسیاری از چالشهای فعلی ژندرمانی را دارد:
- برگشتپذیری (Reversibility): برخلاف CRISPR که تغییرات دائمی ایجاد میکند، اثر ASO موقتی است و اگر عوارض جانبی مشاهده شود، درمان قابل توقف است.
- عدم نیاز به شیمیدرمانی: ژندرمانی فعلی نیازمند دوزهای بالای شیمیدرمانی برای آمادهسازی مغز استخوان است. درمان با ASO میتواند بدون نیاز به این مرحله انجام شود.
- قابلیت دسترسی و مقیاسپذیری: تولید ASOها فرآیندی شیمیایی و ارزانتر از ویرایش سلولی است. این امر میتواند درمان را برای بیماران در کشورهای در حال توسعه قابل دسترس کند.
۵. گزارشهای بالینی ۲۰۲۵: فراتر از آزمایشگاه
سال ۲۰۲۵ شاهد انتشار دادههای حیاتی از کارآزماییهای بالینی در سنت جود بود که اثرات فیزیولوژیک درمانهای مبتنی بر BCL11A را تأیید میکند.
۵.۱ بهبود نوروواسکولار و پیشگیری از سکته مغزی
یکی از خطرناکترین عوارض SCD، سکته مغزی است که ناشی از اختلال در جریان خون مغزی است. در ژوئن ۲۰۲۵، سنت جود نتایج مطالعهای را منتشر کرد که نشان میداد ژندرمانی همودینامیک مغز را نرمال میسازد.
یافتهها: در بیمارانی که تحت ژندرمانی قرار گرفتند، سرعت جریان خون در شریانهای مغزی بین ۲۲٪ تا ۴۳٪ کاهش یافت و به سطوح نرمال رسید. این اثر حفاظتی در پیگیریهای یک و دو ساله پایدار ماند.
۵.۲ کارآزمایی SAGES1 و بهبود کیفیت زندگی
کارآزمایی بالینی SAGES1 که در سنت جود در جریان است، از تکنولوژی CRISPR-Cas9 استفاده میکند. گزارشهای سال ۲۰۲۵ نشان میدهد که شرکتکنندگان بهبودهای چشمگیری در کیفیت زندگی (QoL) تجربه کردهاند. دادهها نشان داد که بیماران در تمام شاخصها از جمله سلامت جسمانی، عملکرد اجتماعی و سلامت عاطفی بهبود یافتهاند.
۶. اهداف ژنتیکی و فناوریهای نوین: فراتر از BCL11A
گزارشهای سال ۲۰۲۵ نشان میدهد که تحقیقات تنها به BCL11A محدود نشده است.
۶.۱ شناسایی هدف جدید: ژن FLT1
در مارس ۲۰۲۵، ژن FLT1 به عنوان یک هدف درمانی جدید معرفی شد. واریانتهای این ژن (پروتئین مرتبط با توسعه عروقی) با سطوح هموگلوبین جنینی مرتبط هستند. ویرایش یا سرکوب FLT1 میتواند به عنوان یک درمان مکمل در کنار سرکوب BCL11A استفاده شود.
۶.۲ ویرایش باز (Base Editing) و ویرایش پرایم
سنت جود همچنان پیشگام توسعه نسل بعدی ابزارهای ویرایش ژنوم است. استفاده از Base Editing (تغییر شیمیایی باز بدون برش DNA) میتواند بسیار دقیقتر و ایمنتر باشد. این روش به جای تخریب، یک جایگاه اتصال جدید برای فاکتور رونویسی TAL1 ایجاد میکند که مستقیماً بیان گاما-گلوبین را فعال میکند.
۶.۳ اپیژنتیک و دمتیلاسیون پروموتر
مطالعه دیگری نقش متیلاسیون DNA در پروموتر گاما-گلوبین را نهایی کرد. حذف متیلاسیون از پروموتر گاما-گلوبین به تنهایی برای فعالسازی مجدد HbF کافی است. این یافته مسیر استفاده از “ویرایشگرهای اپیژنتیک” را باز میکند.
۷. چالشهای انتقال به کلینیک و چشمانداز آینده
۷.۱ چالش تحویل (Delivery) برای درمانهای ASO
اصلیترین مانع برای جایگزینی CRISPR با ASO، رساندن دارو به مغز استخوان است. سنت جود در حال تحقیق بر روی سیستمهای نوین تحویل دارو است، از جمله نانوذرات لیپیدی (LNPs) هدفمند شده و کونژوگههای آنتیبادی-دارو.
۷.۲ تحلیل تکسلولی و هوش مصنوعی
سنت جود در ژانویه ۲۰۲۵ الگوریتم هوش مصنوعی جدیدی به نام CSI-GEP را معرفی کرد. این ابزار امکان تحلیل دادههای بیان ژن در سطح تکسلول را فراهم میکند تا تشخیص دهد آیا ساختار روزت در تکتک سلولهای بیمار تخریب شده است یا خیر.
۷.۳ عدالت در سلامت جهانی
بیمارستان سنت جود تأکید دارد که درمانهای پیشرفته نباید تنها در دسترس بیماران کشورهای ثروتمند باشد. گذار از ژندرمانیهای پیچیده به داروهای قابل تزریق (مانند ASO)، کلید اصلی برای درمان میلیونها بیمار در آفریقا و آسیا است.
۸. نتیجهگیری
گزارش سال ۲۰۲۵ بیمارستان تحقیقاتی کودکان سنت جود، ترسیمکننده آیندهای است که در آن درمان بیماریهای ژنتیکی از “برش کورکورانه DNA” به “مهندسی دقیق معماری ژنوم” تغییر مسیر میدهد. کشف اینکه ژندرمانیهای فعلی با تخریب ساختار روزت کروماتینی عمل میکنند، راهکاری نوین (ASO) را پیش پای محققان گذاشت. ترکیبی از این دانش با پیشرفتهای بالینی، نویدبخش عصری است که در آن کمخونی داسیشکل به عنوان یک شرایط مدیریتشدنی شناخته خواهد شد.
| حوزه تحقیق | نوآوری / کشف کلیدی |
|---|---|
| مکانیسم ژندرمانی | شناسایی نقش ساختار “روزت کروماتینی” و فروپاشی آن در اثر ویرایش |
| هدفگیری مولکولی | کشف نقش حیاتی eRNA و NIPBL در حفظ ساختار سه بعدی |
| روش درمانی نوین | استفاده از ASO برای تخریب eRNA و بازتولید اثر درمانی بدون ویرایش DNA |
| پیامد بالینی (مغز) | نرمالسازی جریان خون مغزی و کاهش ریسک سکته پس از ژندرمانی |
| هدف ژنتیکی جدید | شناسایی ژن FLT1 به عنوان تعدیلکننده هموگلوبین جنینی |
| ابزار تحلیلی | توسعه الگوریتم هوش مصنوعی CSI-GEP برای تحلیل تکسلولی |
این سند بر اساس دادههای موجود تا اواخر سال ۲۰۲۵ تدوین شده و منعکسکننده آخرین وضعیت دانش در حوزه هماتولوژی مولکولی است.
بازبینی توسط متخصص
بازبین علمی این مقاله
آیا در خانواده شما سابقه کمخونی ارثی وجود دارد؟
درمانهای ژنتیکی نوین امیدی تازه برای بیماران مبتلا به تالاسمی و کمخونی داسیشکل ایجاد کردهاند. اگر سوالی در مورد تشخیص ژنتیکی یا گزینههای درمانی جدید دارید، تیم ما آماده ارائه مشاوره تخصصی است.
دریافت مشاوره ژنتیک