
اسکلروز جانبی آمیوتروفیک (ALS)
نقشه یادگیری این مقاله
۱. پادکست: برای درک کلی، ابتدا به پادکست گوش دهید.
۲. ویدیو: ویدیو آموزشی را برای یادگیری عمیق مشاهده کنید.
۳. مطالعه متن: در نهایت، متن مقاله را به عنوان منبع جامع مرور کنید.
مشاهده ویدیو در آپارات
تحلیل جامع اپیدمیولوژیک، ژنتیکی و محیطی بیماری اسکلروز جانبی آمیوتروفیک (ALS)
راهنمای تخصصی برای خانوادههای در معرض خطر
بیماری اسکلروز جانبی آمیوتروفیک (ALS) که در محافل علمی با عنوان بیماری نورونهای حرکتی نیز شناخته میشود، یکی از پیچیدهترین و چالشبرانگیزترین اختلالات نورودژنراتیو در پزشکی مدرن است. این بیماری به طور انتخابی نورونهای حرکتی فوقانی در قشر حرکتی مغز و نورونهای حرکتی تحتانی در ساقه مغز و طناب نخاعی را هدف قرار میدهد. تخریب این سلولهای عصبی حیاتی منجر به قطع ارتباط میان مغز و عضلات ارادی بدن شده، که نتیجه نهایی آن تحلیل رفتن عضلات، فلج تدریجی و در نهایت نارسایی تنفسی است. برای خانوادههایی که با این تشخیص در میان نزدیکان خود مواجه شدهاند یا به دلیل سابقه خانوادگی در معرض خطر قرار دارند، درک عمیق ساختار بیماری، ریسکفاکتورهای محیطی و پیشرفتهای ژنتیکی نه تنها یک ضرورت علمی، بلکه ابزاری برای مدیریت روانی و بالینی آینده است. این گزارش با رویکردی تخصصی به بررسی تفاوتهای ساختاری موارد تکگیر و ارثی، نقش سموم محیطی، تاثیرات خدمت نظامی و دخانیات، و اهمیت حیاتی نشانههای بالینی اولیه میپردازد.
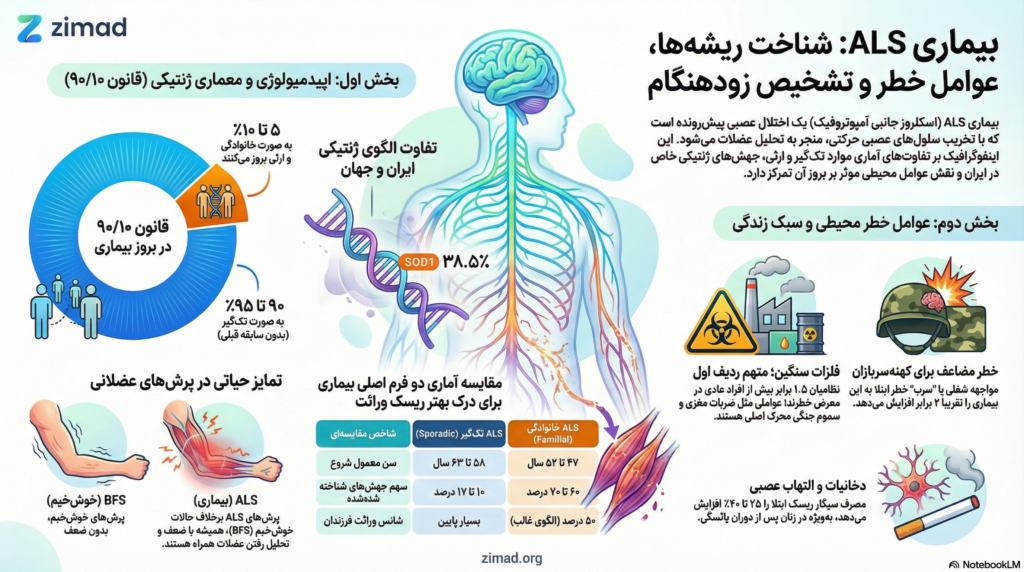
معماری اپیدمیولوژیک ALS: واکاوی قانون ۹۰/۱۰
تقسیمبندی کلاسیک ALS بر اساس الگوی بروز در جمعیت، به دو دسته اصلی “تکگیر” (Sporadic) و “خانوادگی” (Familial) استوار است که در ادبیات علمی به عنوان قانون ۹۰/۱۰ شناخته میشود. این تفکیک اگرچه از نظر بالینی در مراحل پیشرفته بیماری تفاوت چندانی ایجاد نمیکند، اما از منظر سببشناسی (Etiology) و ارزیابی ریسک برای بازماندگان، اهمیت بنیادینی دارد.
موارد تکگیر (Sporadic ALS) و ماهیت چندعاملی آن
تقریباً ۹۰ تا ۹۵ درصد از کل موارد ALS در جهان به صورت تکگیر رخ میدهند، به این معنا که فرد مبتلا هیچ سابقه خانوادگی مشخصی از این بیماری یا اختلالات مرتبط ندارد. در این موارد، بیماری معمولاً در سنین اواخر دهه ۵۰ یا اوایل دهه ۶۰ ظاهر میشود. علیرغم نام “تکگیر”، تحقیقات اخیر نشان دادهاند که این فرم از بیماری به هیچ وجه صرفاً ناشی از شانس نیست؛ بلکه حاصل یک تعامل پیچیده و چند مرحلهای میان استعداد ژنتیکی پنهان و مواجهههای محیطی در طول عمر است.
نکته حائز اهمیت برای خانوادهها این است که فقدان سابقه خانوادگی لزوماً به معنای عدم وجود مولفه ژنتیکی نیست. تخمین زده میشود که حدود ۱۷ درصد از موارد تکگیر در واقع دارای جهشهای ژنتیکی شناخته شدهای هستند که ممکن است به دلیل مرگ زودهنگام والدین (قبل از بروز علائم) یا وقوع جهشهای جدید (De novo) در فرد مبتلا، در شجرهنامه خانواده دیده نشده باشند.
موارد خانوادگی (Familial ALS) و الگوهای وراثت
حدود ۵ تا ۱۰ درصد موارد ALS در دسته خانوادگی قرار میگیرند، که در آن حداقل دو نفر در یک شجرهنامه به این بیماری یا زوال عقل فرونتوتمپورال (FTD) مبتلا شدهاند. در این گروه، علائم معمولاً در سنین پایینتر، یعنی اواخر دهه ۴۰ یا اوایل دهه ۵۰ بروز میکنند. الگوی وراثت در اکثر این خانوادهها “اتوزومال غالب” است؛ بدین معنا که وجود تنها یک کپی از ژن جهشیافته از سوی یکی از والدین برای انتقال خطر بیماری کافی است و هر فرزند ۵۰ درصد شانس ارثبری این جهش را دارد.
با این حال، مفهومی به نام “نفوذ ناقص” (Reduced Penetrance) در این میان وجود دارد که برای خانوادههای در معرض خطر بسیار کلیدی است؛ برخی افراد ممکن است ژن جهشیافته را حمل کنند اما هرگز در طول عمر خود علائم ALS را نشان ندهند. این پدیده نشان میدهد که حتی در موارد صراحتاً ژنتیکی، عوامل دیگری (محیطی یا ژنتیکی محافظتکننده) در تعیین سرنوشت نهایی سلولهای عصبی نقش دارند.
| شاخص مقایسهای | ALS تکگیر (sALS) | ALS خانوادگی (fALS) |
|---|---|---|
| درصد شیوع کل | ۹۰-۹۵٪ | ۵-۱۰٪ |
| سن معمول شروع علائم | ۵۸ تا ۶۳ سال | ۴۷ تا ۵۲ سال |
| سابقه خانوادگی | منفی | مثبت (یک یا چند مورد) |
| سهم جهشهای شناخته شده | ۱۰-۱۷٪ | ۶۰-۷۰٪ |
| ریسک برای فرزندان | مشابه جمعیت عمومی (بسیار پایین) | ۵۰٪ شانس وراثت ژن (در الگوی غالب) |
منظر ژنتیک مولکولی: فراتر از تکرار نوکلئوتیدی
شناسایی ژنهای مسبب ALS نه تنها در تشخیص دقیق، بلکه در ابداع روشهای درمانی “دقیق” (Precision Medicine) انقلابی ایجاد کرده است. بیش از ۴۰ ژن تا به امروز شناسایی شدهاند که جهش در آنها میتواند منجر به نابودی نورونهای حرکتی شود.
جهش در ژن C9orf72 و پیوند با زوال عقل
شایعترین علت ژنتیکی ALS در سطح جهان، انبساط تکرارهای ششنوکلئوتیدی (GGGGCC) در ژن C9orf72 است. در حالی که افراد سالم حدود ۶ تکرار از این توالی را دارند، بیماران ممکن است صدها یا هزاران تکرار داشته باشند. این جهش مسئول ۲۵ تا ۴۰ درصد از موارد خانوادگی و حدود ۶ تا ۷ درصد از موارد تکگیر است. ویژگی منحصربهفرد این جهش، ارتباط مستقیم آن با زوال عقل فرونتوتمپورال (FTD) است؛ به طوری که در یک خانواده، ممکن است یک فرد دچار ناتوانی حرکتی (ALS) شود و فرد دیگر دچار تغییرات شخصیتی و زوال شناختی (FTD) گردد.
ژن SOD1 و اهمیت آن در جامعه ایران
ژن سوپراکسید دیسموتاز ۱ (SOD1) اولین ژنی بود که پیوند آن با ALS در سال ۱۹۹۳ کشف شد. جهشهای این ژن منجر به تاشدگی اشتباه پروتئین و تجمع تودههای سمی در نورونها میشود. نکته بسیار حیاتی برای متخصصان و خانوادههای ایرانی این است که برخلاف جوامع غربی که C9orf72 در آنها شایعتر است، در ایران جهشهای SOD1 نقش بسیار پررنگتری ایفا میکنند. مطالعات بر روی خانوادههای ایرانی نشان داده است که حدود ۱۲ درصد از کل بیماران و ۳۸.۵ درصد از موارد خانوادگی در ایران با جهشهای SOD1 مرتبط هستند، در حالی که سهم C9orf72 در ایران تنها حدود ۲.۶ درصد گزارش شده است. این یافته ضرورت اولویتبندی آزمایشهای ژنتیک برای ژن SOD1 را در خانوادههای ایرانی مبتلا به فرم ارثی دوچندان میکند.
سموم محیطی و فلزات سنگین: تمرکز بر نقش سرب
یکی از بزرگترین دغدغههای خانوادههای در معرض خطر، نقش محیط در تحریک بیماری است. از میان طیف گستردهای از مواد سمی، فلزات سنگین و به ویژه “سرب” به عنوان یکی از قویترین کاندیداهای ریسکفاکتور غیرژنتیکی مطرح شدهاند.
مکانیسمهای سمیت عصبی سرب
سرب یک توکسین عصبی با سابقه طولانی در ایجاد نوروپاتی است. در بیماری ALS، فرضیه بر این است که مواجهه طولانیمدت با سطوح حتی پایین سرب میتواند منجر به تجمع آن در بافتهای عصبی شود. تحقیقات نشان دادهاند که غلظت سرب در مایع مغزی-نخاعی (CSF) و خون بیماران ALS به طور معناداری بالاتر از افراد سالم است. این فلز با تقلید از عملکرد کلسیم، در انتقال پیامهای عصبی اختلال ایجاد کرده و باعث تولید رادیکالهای آزاد و استرس اکسیداتیو شدید در نورونهای حرکتی میشود.
نتایج متاآنالیزهای اپیدمیولوژیک حاکی از آن است که مواجهه شغلی با سرب (مانند کار در صنایع رنگ، باطریسازی، ریختهگری و لولهکشی قدیمی) خطر ابتلا به ALS را تقریباً دو برابر (OR = 1.81) میکند.
| عامل محیطی | نسبت شانس (OR) / خطر نسبی (RR) | تفسیر ریسک |
|---|---|---|
| مواجهه با سرب (Occupational Lead) | ۱.۸۱ تا ۲.۳۱ | افزایش حدود ۱۰۰ درصدی خطر |
| آفتکشها (Pesticides) | حدود ۲.۰ | افزایش دو برابری خطر |
| فلزات سنگین (General Metals) | ۱.۴۵ | افزایش ۴۵ درصدی خطر |
| حلالهای شیمیایی (Solvents) | متغیر | ارتباط ضعیف تا متوسط |
این دادهها نشاندهنده اهمیت “بهداشت حرفهای” برای افرادی است که سابقه خانوادگی ALS دارند؛ کاهش مواجهه با آلایندههای صنعتی میتواند یک لایه محافظتی مهم در برابر فعال شدن مسیرهای تخریب عصبی باشد.
خدمت سربازی و ALS: معمای اپیدمیولوژیک
یکی از عجیبترین و در عین حال مستندترین یافتهها در تحقیقات ALS، نرخ بالای ابتلای کهنهسربازان نظامی به این بیماری است. مطالعات متعددی نشان دادهاند که افرادی که در نیروهای مسلح خدمت کردهاند، فارغ از شاخه نظامی، دوران خدمت (جنگ یا صلح) و منطقه جغرافیایی، با ریسک بیشتری برای تشخیص ALS روبرو هستند.
شواهد و علل احتمالی در محیطهای نظامی
تحقیقات بر روی سربازان جنگ خلیج فارس اولین جرقهها را در این زمینه ایجاد کرد و نشان داد که نرخ ابتلای آنها دو برابر جمعیت عادی است. مطالعات بعدی بر روی کهنهسربازان جنگ جهانی دوم و کره نیز نتایج مشابهی را نشان داد. اخیراً در سال ۲۰۲۵، گزارشهایی منتشر شده که نشان میدهد حتی سربازان دوران پس از ۱۱ سپتامبر نیز با نرخهای بالاتری از این بیماری دست و پنجه نرم میکنند.
دلایل پیشنهادی برای این پدیده شامل موارد زیر است:
- مواجهه با مواد سمی خاص: دود ناشی از سوختن چاههای نفت، گازهای شیمیایی، و استفاده گسترده از حلالهای صنعتی و سوخت دیزل در ماشینآلات نظامی.
- تروما و ضربات مغزی: آسیبهای مغزی ضربهای (TBI) که در محیطهای نظامی شایع است، به عنوان یک محرک قوی برای شروع فرآیندهای تخریبی پروتئین TDP-43 (پروتئین کلیدی در ALS) شناخته میشود.
- استرس فیزیکی شدید: فعالیتهای بدنی فرساینده و فراتر از ظرفیت فیزیولوژیک بدن که ممکن است منجر به ناپایداری در متابولیسم نورونهای حرکتی شود.
به دلیل قدرت این شواهد، سازمانهای حمایتی مانند وزارت امور کهنهسربازان آمریکا (VA)، بیماری ALS را به عنوان یک بیماری “صد در صد مرتبط با خدمت” تلقی میکنند.
دخانیات و نیکوتین: ریسکفاکتور قابل اصلاح
مصرف سیگار به عنوان یکی از جدیترین عوامل خطر محیطی که توسط فرد قابل کنترل است، در کانون توجه مطالعات پیشگیرانه قرار دارد. اگرچه در برخی مطالعات نتایج متناقضی مشاهده شده، اما تجمیع دادههای حاصل از متاآنالیزهای بزرگ (۲۰۲۳-۲۰۲۴) نشاندهنده یک رابطه مثبت میان مصرف دخانیات و خطر ALS است.
مکانیسمهای آسیبرسان سیگار و تفاوتهای جنسیتی
سیگار از چندین مسیر به سیستم عصبی آسیب میزند:
- استرس اکسیداتیو: دود سیگار حاوی رادیکالهای آزاد بیشماری است که باعث اکسیداسیون لیپیدها و پروتئینهای سلولی میشود.
- التهاب عصبی (Neuroinflammation): مواد موجود در تنباکو باعث فعال شدن غیرطبیعی میکروگلیاها و آستروسیتها در مغز شده و منجر به آزادسازی سیتوکینهای التهابی مانند TNF-α و IFN-γ میگردد.
- فلزات سمی: سیگار منبع مواجهه با فلزاتی نظیر کادمیوم و سرب است که پیشتر به سمیت آنها اشاره شد.
یافتههای اپیدمیولوژیک نشان میدهند که سیگار کشیدن خطر ابتلا را حدود ۲۵ تا ۴۰ درصد افزایش میدهد. نکته جالب توجه این است که این خطر در زنان سیگاری، به ویژه پس از دوران یائسگی، به طور معناداری بالاتر از مردان گزارش شده است.
فاسیکولاسیونها: تشخیص زودهنگام و تمایز حیاتی
یکی از چالشبرانگیزترین لحظات برای خانوادههای در معرض خطر، مشاهده پرشهای عضلانی ناگهانی یا همان “فاسیکولاسیون” است. این لرزشهای ریز زیرپوستی میتوانند در افراد کاملاً سالم نیز رخ دهند، اما در زمینه ALS، آنها اغلب نخستین نشانههای فیزیکی از تخریب واحد حرکتی هستند.
تفاوتهای بالینی فاسیکولاسیونهای ALS و حالات خوشخیم
سندرم فاسیکولاسیون خوشخیم (BFS) وضعیتی است که در آن فرد دچار پرشهای عضلانی مداوم میشود اما هیچ بیماری عصبی زمینهای ندارد. برای خانوادهها بسیار مهم است که بدانند فاسیکولاسیون به تنهایی تشخیصدهنده ALS نیست؛ پزشک تنها زمانی به ALS مشکوک میشود که این پرشها با ضعف عضلانی پیشرونده و تحلیل رفتن عضلات (آتروفی) همراه باشند.
| ویژگی افتراقی | فاسیکولاسیون در ALS (بدخیم) | فاسیکولاسیون در BFS (خوشخیم) |
|---|---|---|
| همراهی با ضعف عضلانی | تقریباً همیشه وجود دارد | هرگز وجود ندارد |
| آتروفی (تحلیل عضله) | شایع و پیشرونده | وجود ندارد |
| محل شروع و توزیع | اغلب در چندین اندام همزمان؛ تمایل به نواحی نزدیک تنه (مانند شانه) | اغلب محدود به یک نقطه (مانند پلک یا ساق پا) |
| فرکانس شلیک در EMG | نرخ شلیک بسیار بالا و نامنظم | نرخ شلیک پایینتر و منظمتر |
| ویژگیهای الکتریکی | همراه با پتانسیلهای غیرطبیعی (Denervation) | نوار عصب و عضله در سایر موارد نرمال است |
| عوامل تحریککننده | تخریب بیولوژیک اعصاب | استرس، خستگی، کافئین، اضطراب |
مدیریت و پیشگیری در خانوادههای پرخطر: راهنمای عملی
برای خانوادههایی که یک مورد ارثی (fALS) در آنها تایید شده است، مدیریت آینده نیازمند ترکیبی از پایش علمی و حمایتهای روانی-اجتماعی است.
آزمایش ژنتیک و ملاحظات اخلاقی-قانونی
آزمایش ژنتیک برای افراد سالم خانواده (Predictive Testing) یک انتخاب شخصی است که باید حتماً تحت نظر “مشاور ژنتیک” انجام شود. این آزمایش میتواند بار روانی سنگینی داشته باشد و بر مسائلی نظیر بیمه عمر یا برنامهریزی برای فرزندآوری تاثیر بگذارد. با این حال، شناسایی زودهنگام ناقلین جهشهای خاص مانند SOD1 اکنون اهمیت درمانی پیدا کرده است، زیرا داروهای جدیدی در حال آزمایش هستند که هدف آنها جلوگیری از شروع علائم در ناقلین پیشعلامتی است.
نشانگرهای زیستی: افقهای روشن در پایش
امروزه پایش افراد در معرض خطر از طریق سنجش سطح “زنجیره سبک نوروفیلامنت” (NfL) در خون انجام میشود. این پروتئین زمانی که فیبرهای عصبی آسیب میبینند، در خون آزاد میشود. در افراد ناقل جهش، افزایش ناگهانی NfL میتواند نشانهای از شروع قریبالوقوع بیماری (حتی قبل از بروز لرزش عضله) باشد، که اجازه میدهد درمانهای محافظتی سریعاً آغاز گردند.
استراتژیهای کاهش خطر
اگرچه تغییر ژنتیک ممکن نیست، اما خانوادههای در معرض خطر میتوانند با اقدامات زیر، “آستانه” بروز بیماری را جابجا کنند:
- حذف کامل دخانیات: برای جلوگیری از التهاب عصبی سمی.
- محافظت در برابر فلزات سنگین: اطمینان از سلامت آب مصرفی (فقدان سرب) و استفاده از تجهیزات حفاظتی در مشاغل فنی.
- کنترل ضربات مغزی: استفاده از کلاه ایمنی در ورزشهای تماسی و دوچرخهسواری.
- حمایتهای نهادی: در ایران، پیوستن به “جمعیت حمایت از بیماران ایالاس و نوروپاتی ایران” میتواند دسترسی به آخرین یافتههای پژوهشی و مشاورههای متخصصین داخلی را تسهیل کند.
جمعبندی و چشمانداز آینده
بیماری اسکلروز جانبی آمیوتروفیک، علیرغم ماهیت تهاجمی خود، امروزه بیش از هر زمان دیگری در تاریخ پزشکی شناخته شده است. تمایز میان قانون ۹۰/۱۰ به ما میآموزد که اکثر موارد ناشی از ترکیبی از شانس محیطی و زمینههای ژنتیکی ظریف هستند، در حالی که موارد ارثی، مسیرهای مشخصی را برای درمانهای هدفمند نشان میدهند. نقش غیرقابل انکار سرب، دخانیات و مخاطرات نظامی، بر این واقعیت تاکید دارد که ALS صرفاً یک سرنوشت محتوم ژنتیکی نیست، بلکه محصول تعامل بدن ما با جهان پیرامون است.
شناسایی زودهنگام فاسیکولاسیونها و افتراق آنها از موارد خوشخیم، اولین خط دفاعی در مسیر درمان است. با پیشرفتهای خیرهکننده در حوزه ژندرمانی و شناسایی نشانگرهای زیستی نظیر NfL، امید به تغییر مسیر زندگی افراد در معرض خطر از یک احتمال دور به یک واقعیت نزدیک بالینی تبدیل شده است. خانوادهها با آگاهی از این جزئیات، نه تنها میتوانند با آمادگی بیشتری با بیماری روبرو شوند، بلکه میتوانند فعالانه در کاهش ریسکهای محیطی و حمایت از نسلهای آینده گام بردارند.
بازبینی توسط متخصص
بازبین علمی این مقاله
آیا نگران سابقه خانوادگی ALS هستید؟
اگر در خانواده خود سابقه بیماری ALS دارید، انجام آزمایش ژنتیک و مشاوره تخصصی میتواند به شما در درک ریسکها و اقدامات پیشگیرانه کمک کند.
دریافت مشاوره ژنتیک